

Host Genetics and Environment Shape the Composition of the Gastrointestinal Microbiome in Nonhuman Primates
- PMID: 36475838
- PMCID: PMC9927375
- DOI: 10.1128/spectrum.02139-22
Host Genetics and Environment Shape the Composition of the Gastrointestinal Microbiome in Nonhuman Primates
Abstract
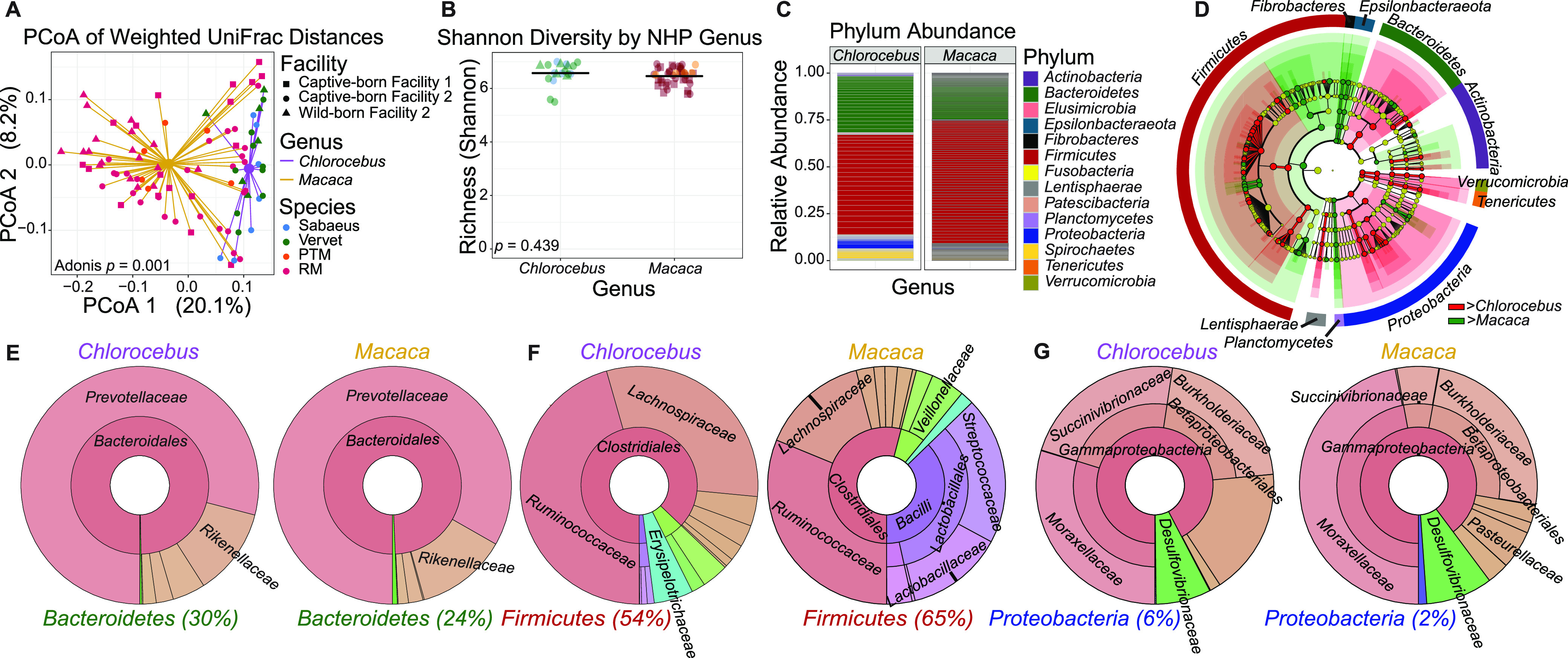
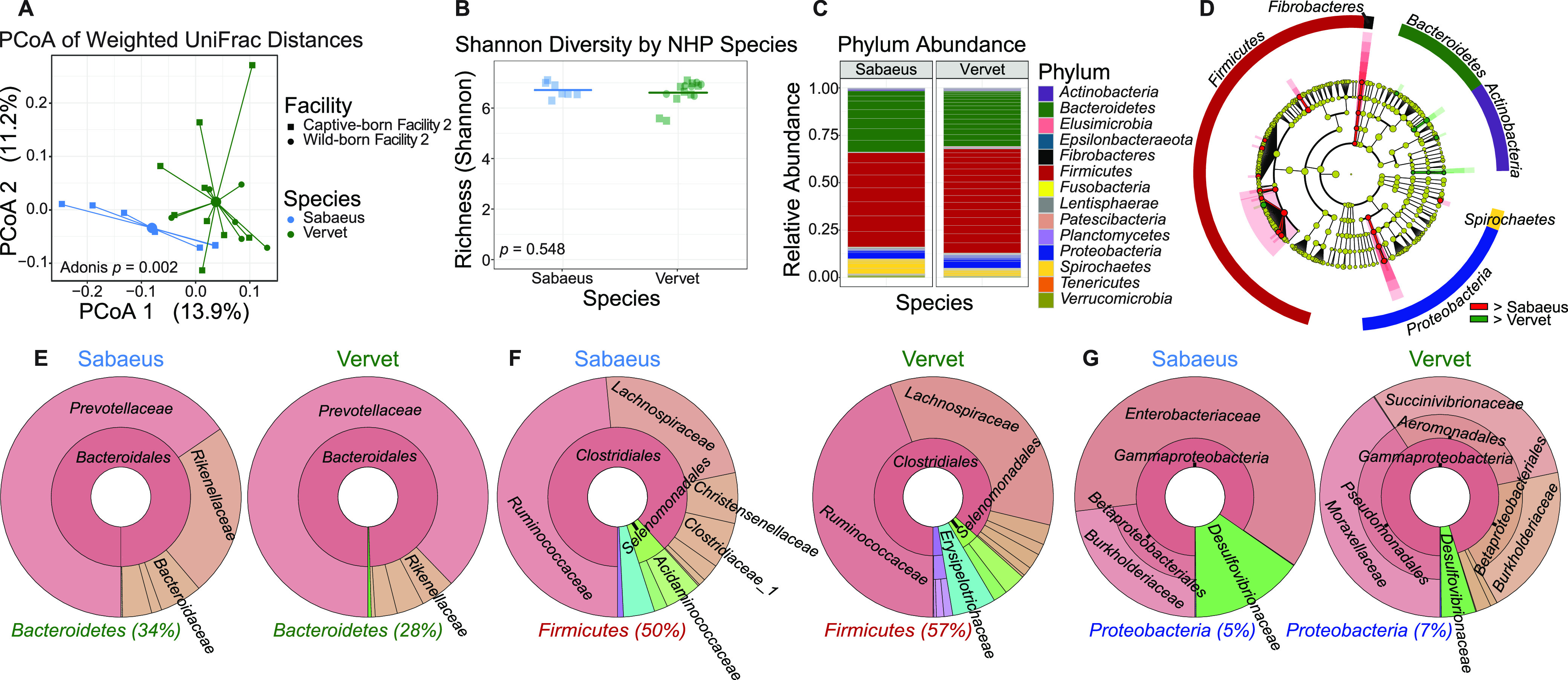
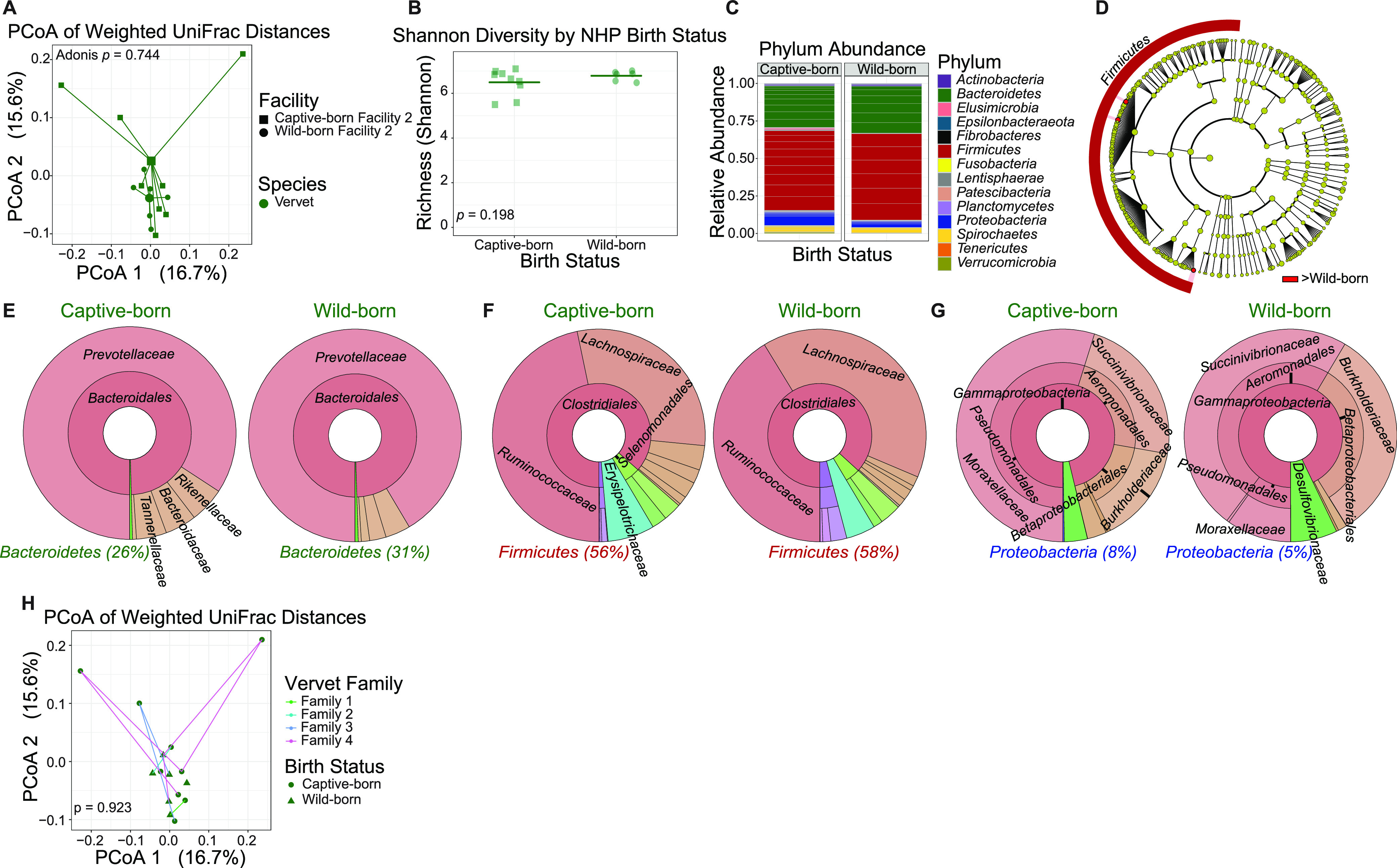
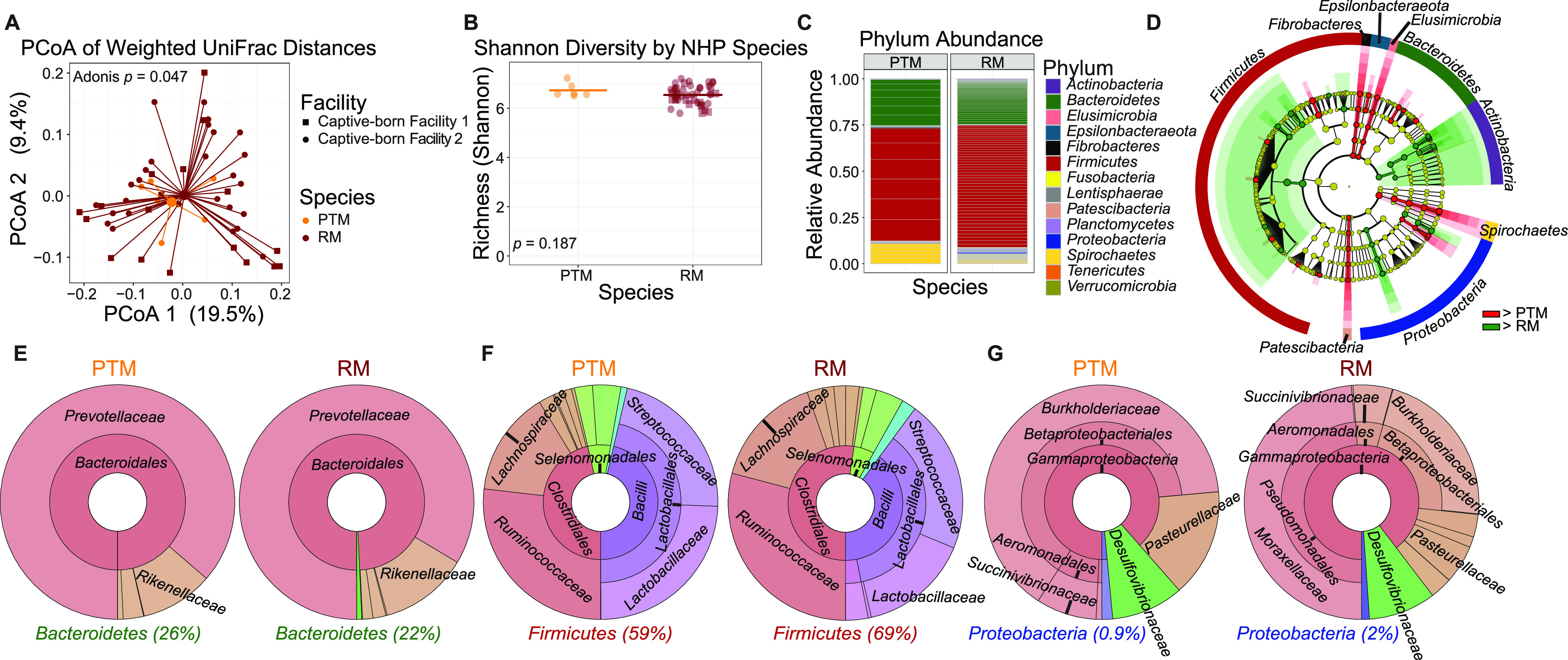
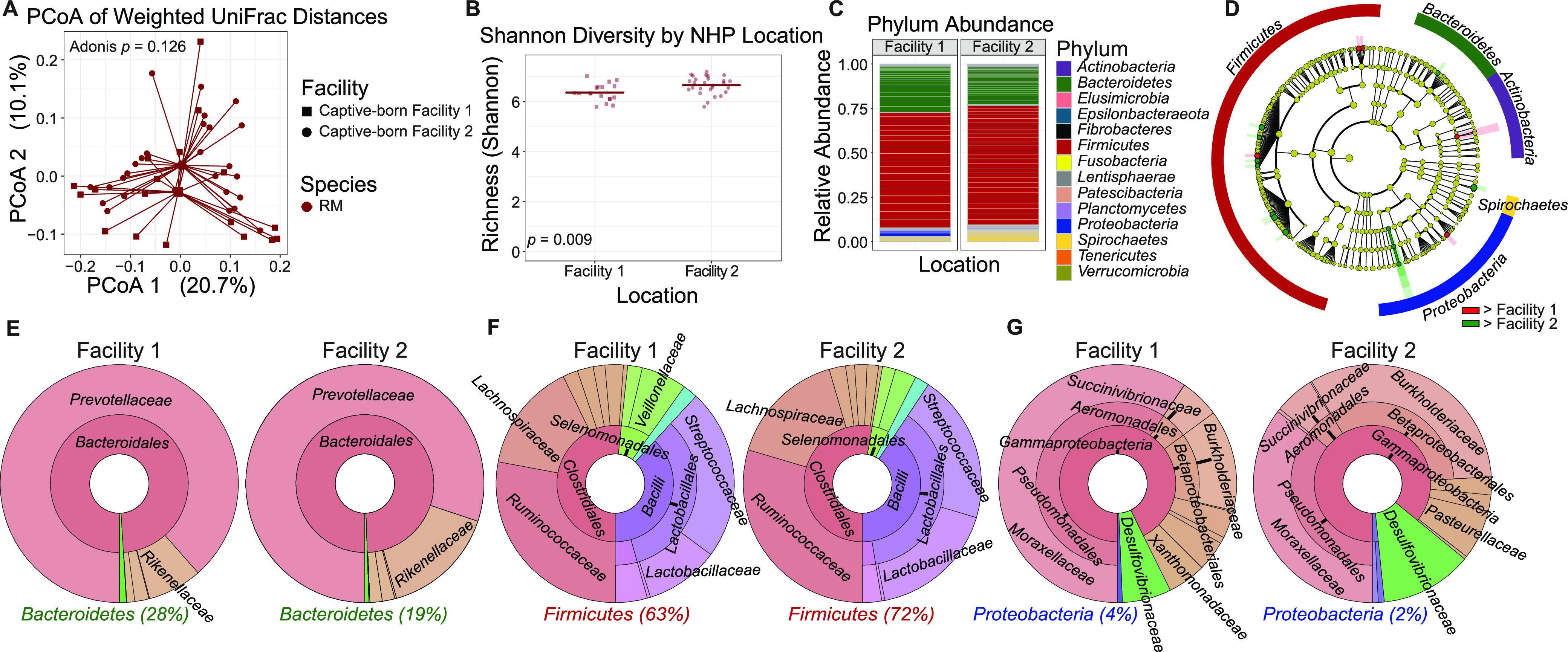
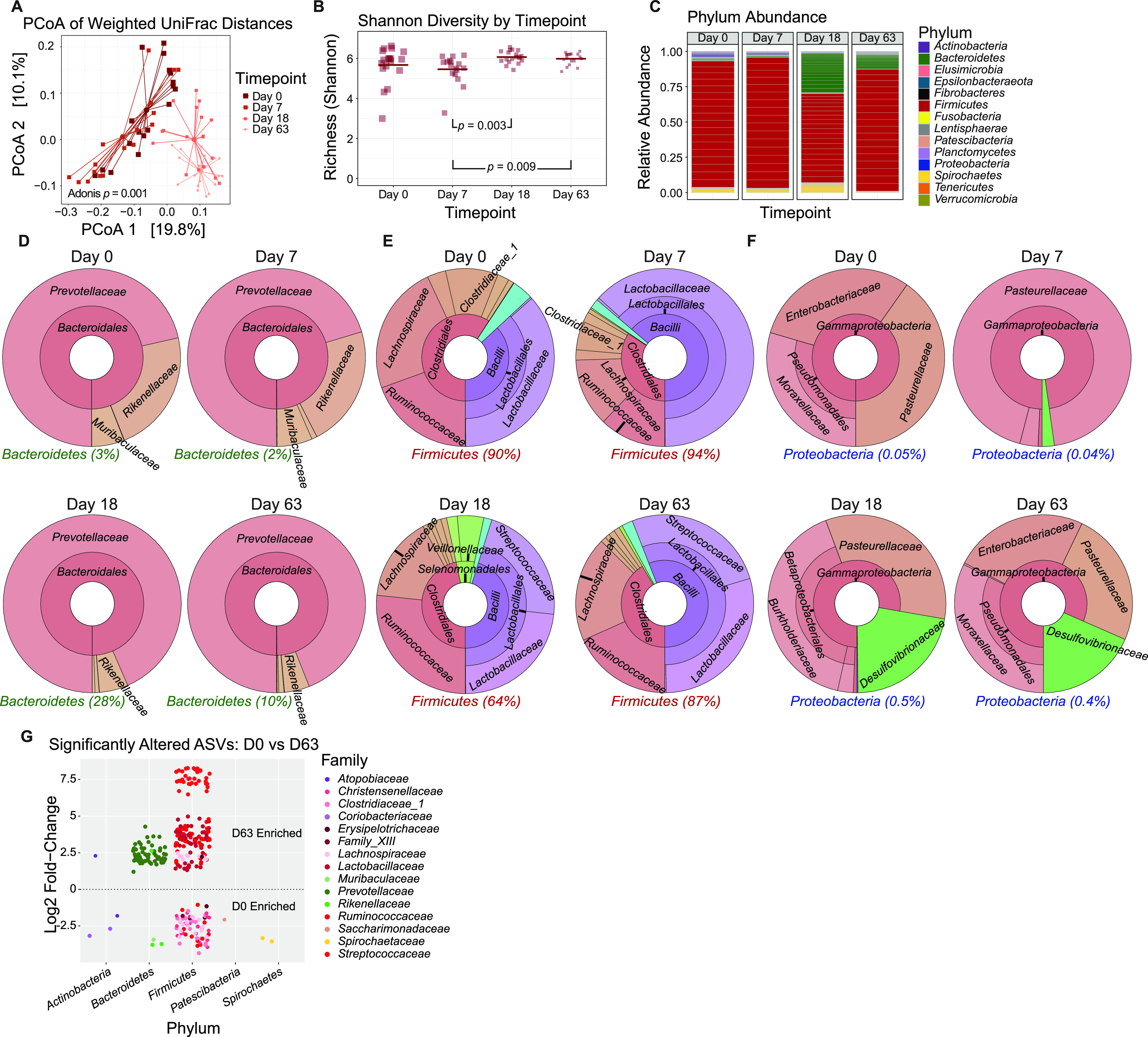
The bacterial component of the gastrointestinal tract microbiome is comprised of hundreds of species, the majority of which live in symbiosis with the host. The bacterial microbiome is influenced by host diet and disease history, and host genetics may additionally play a role. To understand the degree to which host genetics shapes the gastrointestinal tract microbiome, we studied fecal microbiomes in 4 species of nonhuman primates (NHPs) held in separate facilities but fed the same base diet. These animals include Chlorocebus pygerythrus, Chlorocebus sabaeus, Macaca mulatta, and Macaca nemestrina. We also followed gastrointestinal tract microbiome composition in 20 Macaca mulatta (rhesus macaques [RMs]) as they transitioned from an outdoor to indoor environment and compared 6 Chlorocebus pygerythrus monkeys that made the outdoor to indoor transition to their 9 captive-born offspring. We found that genetics can influence microbiome composition, with animals of different genera (Chlorocebus versus Macaca) having significantly different gastrointestinal (GI) microbiomes despite controlled diets. Animals within the same genera have more similar microbiomes, although still significantly different, and animals within the same species have even more similar compositions that are not significantly different. Significant differences were also not observed between wild-born and captive-born Chlorocebus pygerythrus, while there were significant changes in RMs as they transitioned into captivity. Together, these results suggest that the effects of captivity have a larger impact on the microbiome than other factors we examined within a single NHP species, although host genetics does significantly influence microbiome composition between NHP genera and species. IMPORTANCE Our data point to the degree to which host genetics can influence GI microbiome composition and suggest, within primate species, that individual host genetics is unlikely to significantly alter the microbiome. These data are important for the development of therapeutics aimed at altering the microbiome within populations of genetically disparate members of primate species.
Keywords: GI tract microbiome; host genetics; nonhuman primates.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
