

Extensive Recombination-driven Coronavirus Diversification Expands the Pool of Potential Pandemic Pathogens
- PMID: 36477201
- PMCID: PMC9730504
- DOI: 10.1093/gbe/evac161
Extensive Recombination-driven Coronavirus Diversification Expands the Pool of Potential Pandemic Pathogens
Abstract
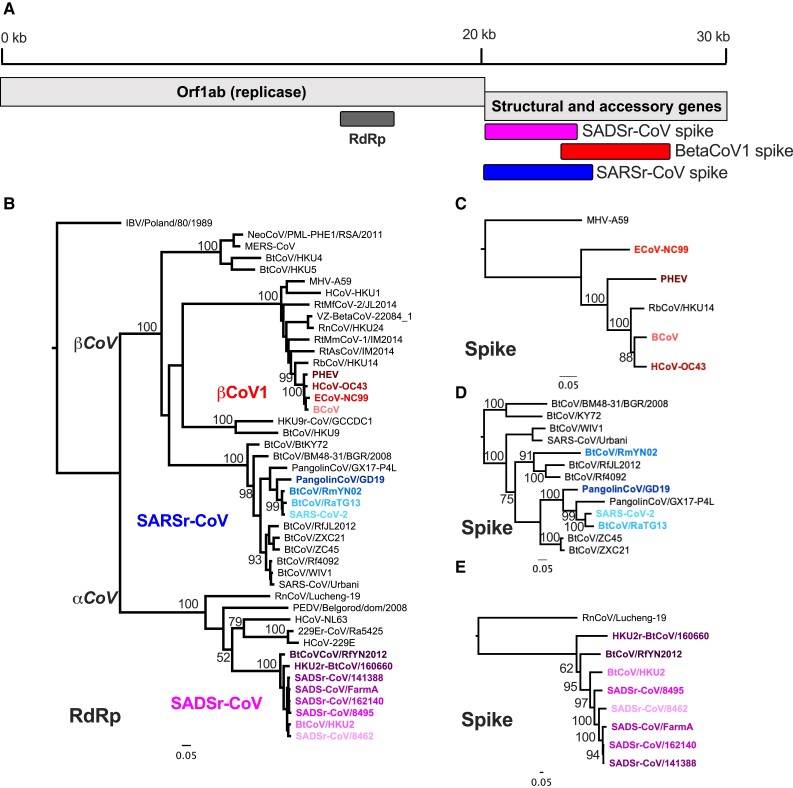
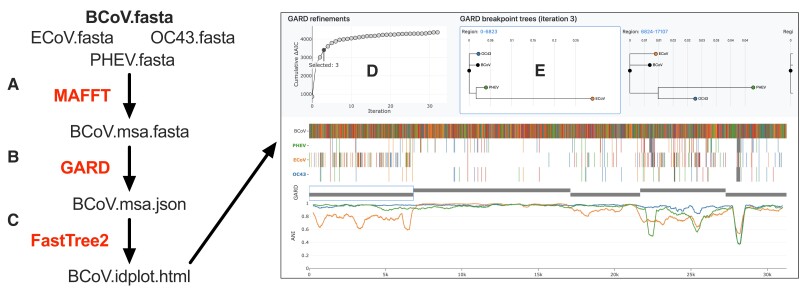
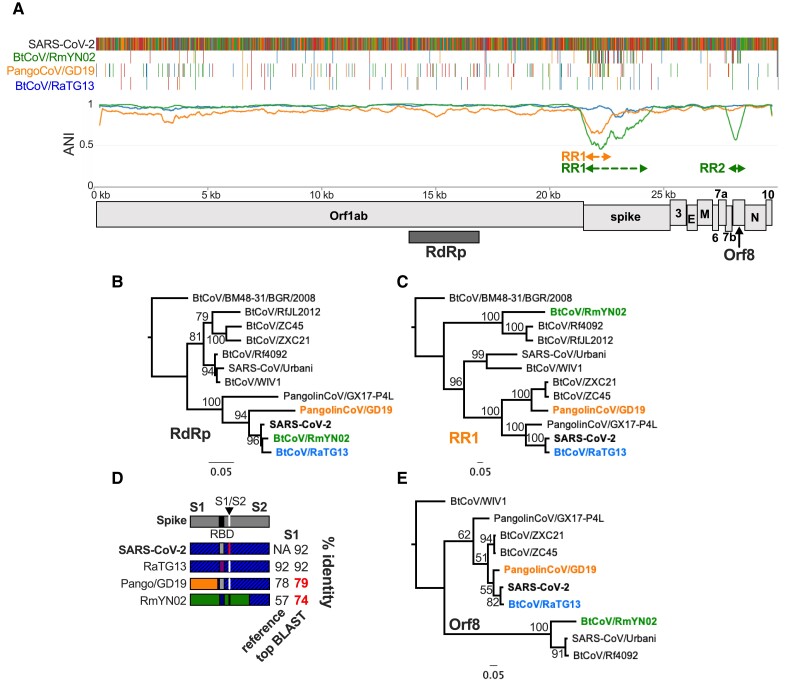
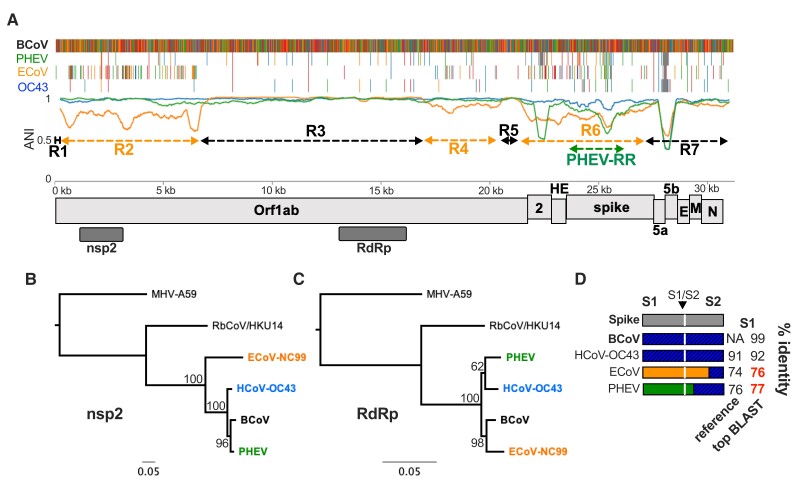
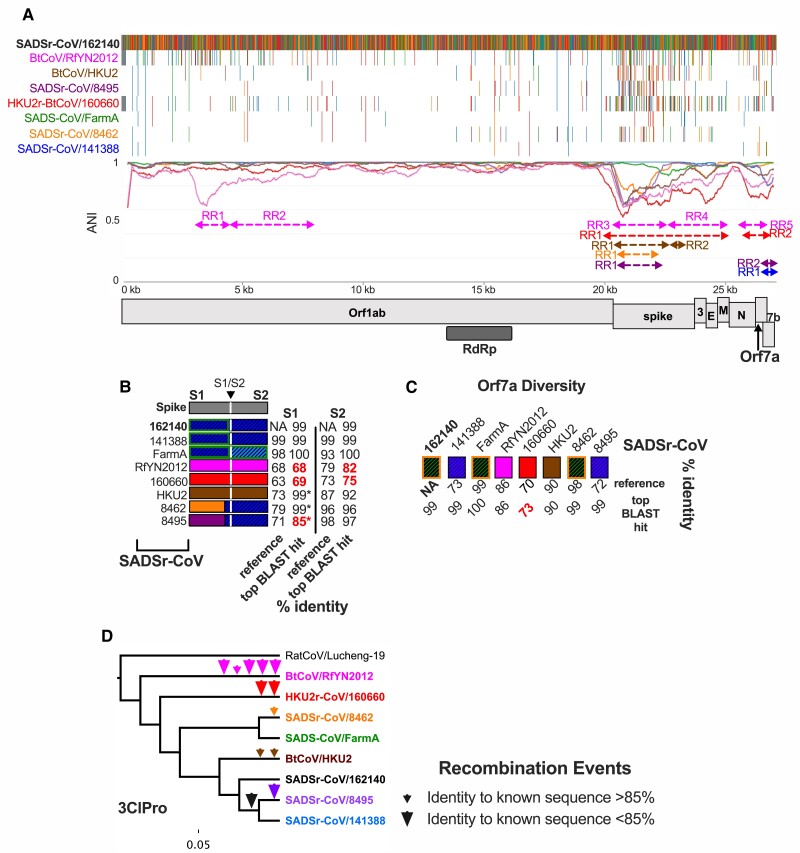
The ongoing SARS-CoV-2 pandemic is the third zoonotic coronavirus identified in the last 20 years. Enzootic and epizootic coronaviruses of diverse lineages also pose a significant threat to livestock, as most recently observed for virulent strains of porcine epidemic diarrhea virus (PEDV) and swine acute diarrhea-associated coronavirus (SADS-CoV). Unique to RNA viruses, coronaviruses encode a proofreading exonuclease (ExoN) that lowers point mutation rates to increase the viability of large RNA virus genomes, which comes with the cost of limiting virus adaptation via point mutation. This limitation can be overcome by high rates of recombination that facilitate rapid increases in genetic diversification. To compare the dynamics of recombination between related sequences, we developed an open-source computational workflow (IDPlot) that bundles nucleotide identity, recombination, and phylogenetic analysis into a single pipeline. We analyzed recombination dynamics among three groups of coronaviruses with noteworthy impacts on human health and agriculture: SARSr-CoV, Betacoronavirus-1, and SADSr-CoV. We found that all three groups undergo recombination with highly diverged viruses from undersampled or unsampled lineages, including in typically highly conserved regions of the genome. In several cases, no parental origin of recombinant regions could be found in genetic databases, demonstrating our shallow characterization of coronavirus diversity and expanding the genetic pool that may contribute to future zoonotic events. Our results also illustrate the limitations of current sampling approaches for anticipating zoonotic threats to human and animal health.
Keywords: coronaviruses; evolution; virology.
© The Author(s) 2022. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
Update of
-
Extensive recombination-driven coronavirus diversification expands the pool of potential pandemic pathogens.bioRxiv [Preprint]. 2021 Jun 28:2021.02.03.429646. doi: 10.1101/2021.02.03.429646. bioRxiv. 2021. Update in: Genome Biol Evol. 2022 Dec 8;14(12):evac161. doi: 10.1093/gbe/evac161. PMID: 33564759 Free PMC article. Updated. Preprint.
References
-
- Boni MF, et al. . 2020. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 5:1408–1417. - PubMed
-
- Debat HJ. 2018. Expanding the size limit of RNA viruses: evidence of a novel divergent nidovirus in California sea hare, with a ∼35.9 kb virus genome. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory.
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
