

Structural variants drive context-dependent oncogene activation in cancer
- PMID: 36477537
- PMCID: PMC9810360
- DOI: 10.1038/s41586-022-05504-4
Structural variants drive context-dependent oncogene activation in cancer
Abstract
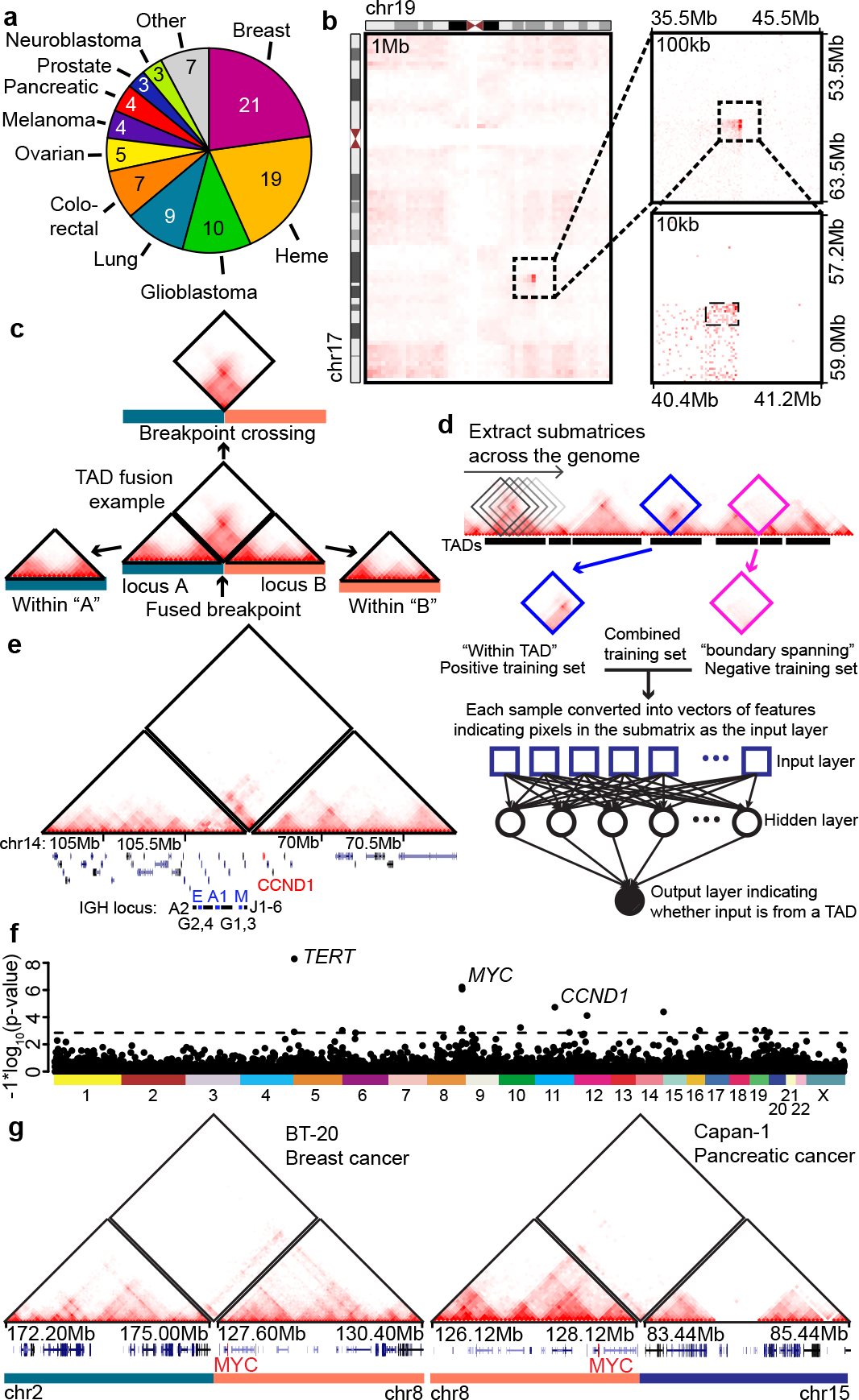
Higher-order chromatin structure is important for the regulation of genes by distal regulatory sequences1,2. Structural variants (SVs) that alter three-dimensional (3D) genome organization can lead to enhancer-promoter rewiring and human disease, particularly in the context of cancer3. However, only a small minority of SVs are associated with altered gene expression4,5, and it remains unclear why certain SVs lead to changes in distal gene expression and others do not. To address these questions, we used a combination of genomic profiling and genome engineering to identify sites of recurrent changes in 3D genome structure in cancer and determine the effects of specific rearrangements on oncogene activation. By analysing Hi-C data from 92 cancer cell lines and patient samples, we identified loci affected by recurrent alterations to 3D genome structure, including oncogenes such as MYC, TERT and CCND1. By using CRISPR-Cas9 genome engineering to generate de novo SVs, we show that oncogene activity can be predicted by using 'activity-by-contact' models that consider partner region chromatin contacts and enhancer activity. However, activity-by-contact models are only predictive of specific subsets of genes in the genome, suggesting that different classes of genes engage in distinct modes of regulation by distal regulatory elements. These results indicate that SVs that alter 3D genome organization are widespread in cancer genomes and begin to illustrate predictive rules for the consequences of SVs on oncogene activation.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Declaration of Interests
The authors declare no competing financial interests
Figures










References
Methods References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
