

[Main characteristics of the new authorisation procedure for clinical trials with medicinal products according to regulation (EU) no 536/2014 and the cooperation between the member states]
- PMID: 36478279
- PMCID: PMC9832078
- DOI: 10.1007/s00103-022-03621-z
[Main characteristics of the new authorisation procedure for clinical trials with medicinal products according to regulation (EU) no 536/2014 and the cooperation between the member states]
Abstract
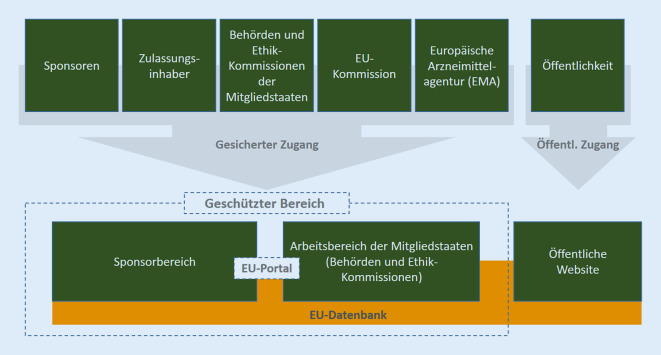
With Regulation (EU) No. 536/2014 on clinical trials on medicinal products for human use, which became applicable on 31 January 2022, full harmonisation of the authorisation and monitoring procedures of clinical trials with medicinal products in the European Union (EU) and the European Economic Area (EEA) has been achieved. In addition to an entirely paperless application procedure, communication between all parties involved is done through the Clinical Trials Information System (CTIS), which was developed specifically for the Regulation and through which all non-proprietary information and content of the clinical trial application and results are also made available to the public. As was already the case under the old legal framework, the authorisation of a clinical trial is granted by each Member State concerned; however, the assessment of the common part of the dossier of a clinical trial that is conducted in more than one Member State is jointly done by the respective Member States under the coordinating lead of a reporting Member State. The present article outlines the authorisation procedure with its deadline concept and addresses further aspects of the Regulation, such as details on the protection of the trial subjects, safety reporting and transparency rules.
Mit der am 31.01.2022 anwendbar gewordenen Verordnung (EU) Nr. 536/2014 zu klinischen Prüfungen mit Humanarzneimitteln wurde die weitgehende Vollharmonisierung der Genehmigungs- und Überwachungsverfahren klinischer Arzneimittelprüfungen in der Europäischen Union (EU) und dem Europäischen Wirtschaftsraum (EWR) vollzogen. Neben einem vollständig papierlosen Antragsverfahren erfolgt auch die gesamte Kommunikation aller Beteiligten über das eigens für die Verordnung entwickelte Clinical Trials Information System (CTIS), über das auch – jeweils zeitlich gestaffelt – alle nicht geschützten Informationen und Inhalte des Genehmigungsantrags und der Ergebnisse der klinischen Prüfung der Öffentlichkeit zugänglich gemacht werden. Wie bereits unter den alten rechtlichen Rahmenbedingungen ergeht die Genehmigung einer klinischen Prüfung durch die jeweils betroffenen Mitgliedstaaten. In den Fällen, in denen eine klinische Prüfung in mehreren Mitgliedstaaten durchgeführt werden soll, erfolgt die Bewertung des allgemeinen Teils der Unterlagen nunmehr gemeinsam durch die betroffenen Mitgliedstaaten unter koordinierender Federführung eines berichterstattenden Mitgliedstaates. Der vorliegende Artikel skizziert das Genehmigungsverfahren mit seinem Fristenkonzept und adressiert weitere Aspekte der Verordnung, wie z. B. Details zum Schutz der an der klinischen Prüfung teilnehmenden Personen, die Sicherheitsberichterstattung sowie die Transparenzregelungen.
Keywords: Clinical Trials Information System (CTIS); Clinical Trials Regulation (CTR); Clinical trials on human medicinal products; Ethics committees; Pharmacovigilance.
© 2022. The Author(s).
References
-
- (2014) Verordnung (EU) Nr. 536/2014 des Europäischen Parlaments und des Rates vom 16. April 2014 über klinische Prüfungen mit Humanarzneimitteln und zur Aufhebung der Richtlinie 2001/20/EG. ABl. L 158, 27.05.2014, S. 1–76
-
- European Medicines Agency (2020) CTIS Highlights. https://www.ema.europa.eu/en/documents/newsletter/clinical-trials-inform.... Zugegriffen: 5. Aug. 2022
-
- (2022) Clinical Trials Information System. https://www.ema.europa.eu/en/human-regulatory/research-development/clini.... Zugegriffen: 5. Aug. 2022
-
- (2001) Richtlinie 2001/20/EG des europäischen Parlaments und des Rates vom 4. April 2001 zur Angleichung der Rechts- und Verwaltungsvorschriften der Mitgliedstaaten über die Anwendung der guten klinischen Praxis bei der Durchführung von klinischen Prüfungen mit Humanarzneimitteln. ABl. L 121, 1.5.2001, S. 34
-
- (2022) Gesetz über den Verkehr mit Arzneimitteln (Arzneimittelgesetz – AMG); neugefasst durch B. v. 12. Dez. 2005 BGBl. I S. 3394; zuletzt geändert durch Artikel 14 G. v. 24. Juni 2022 BGBl. I S. 959
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
