

Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction
- PMID: 36480284
- PMCID: PMC9927948
- DOI: 10.1172/JCI159498
Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction
Abstract
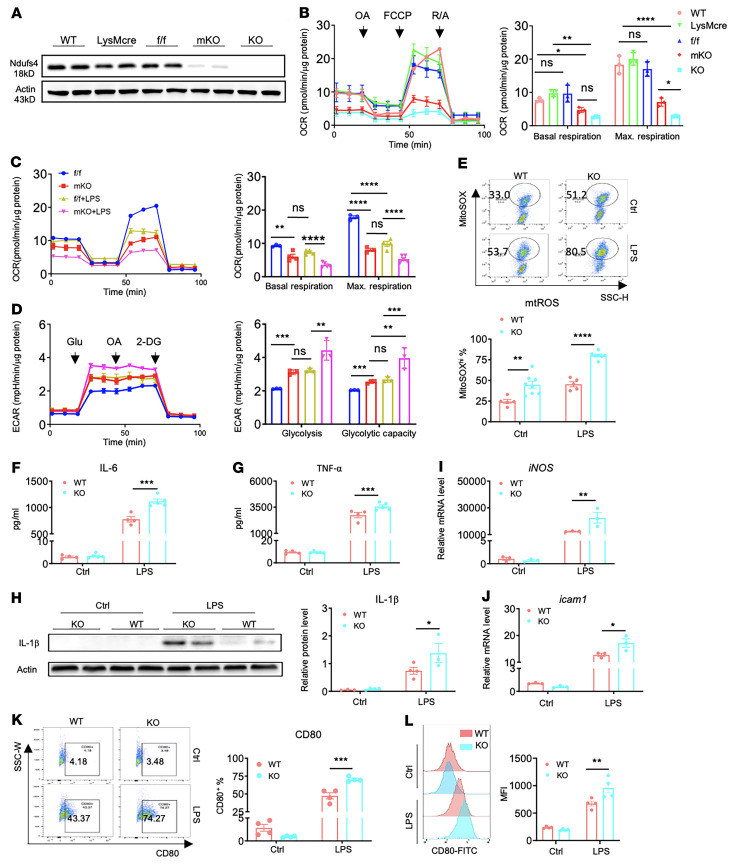
Innate immune cells play important roles in tissue injury and repair following acute myocardial infarction (MI). Although reprogramming of macrophage metabolism has been observed during inflammation and resolution phases, the mechanistic link to macrophage phenotype is not fully understood. In this study, we found that myeloid-specific deletion (mKO) of mitochondrial complex I protein, encoded by Ndufs4, reproduced the proinflammatory metabolic profile in macrophages and exaggerated the response to LPS. Moreover, mKO mice showed increased mortality, poor scar formation, and worsened cardiac function 30 days after MI. We observed a greater inflammatory response in mKO mice on day 1 followed by increased cell death of infiltrating macrophages and blunted transition to the reparative phase during post-MI days 3-7. Efferocytosis was impaired in mKO macrophages, leading to lower expression of antiinflammatory cytokines and tissue repair factors, which suppressed the proliferation and activation of myofibroblasts in the infarcted area. Mitochondria-targeted ROS scavenging rescued these impairments, improved myofibroblast function in vivo, and reduced post-MI mortality in mKO mice. Together these results reveal a critical role of mitochondria in inflammation resolution and tissue repair via modulation of efferocytosis and crosstalk with fibroblasts. These findings have potential significance for post-MI recovery as well as for other inflammatory conditions.
Keywords: Cardiology; Cardiovascular disease; Macrophages; Metabolism; Mitochondria.
Figures





Comment in
- Mitochondrial function in macrophages controls cardiac repair after myocardial infarction doi: 10.1172/JCI167079
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
