

Biophysical characterization of calcium-binding and modulatory-domain dynamics in a pentameric ligand-gated ion channel
- PMID: 36480474
- PMCID: PMC9897478
- DOI: 10.1073/pnas.2210669119
Biophysical characterization of calcium-binding and modulatory-domain dynamics in a pentameric ligand-gated ion channel
Abstract
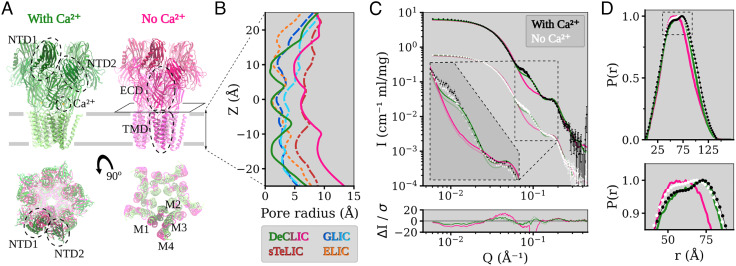
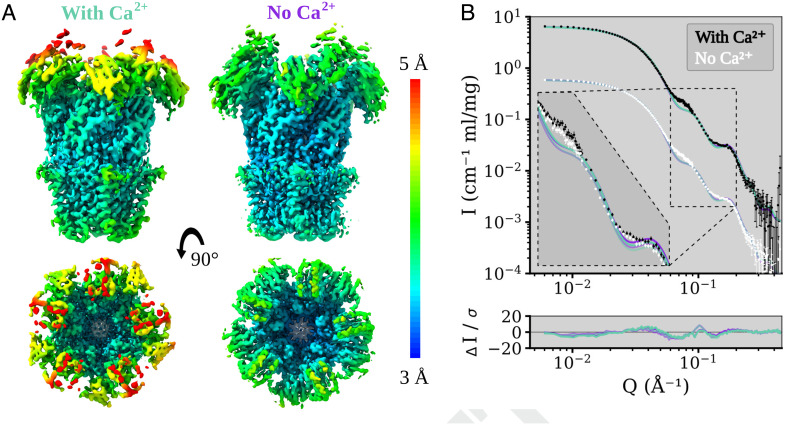
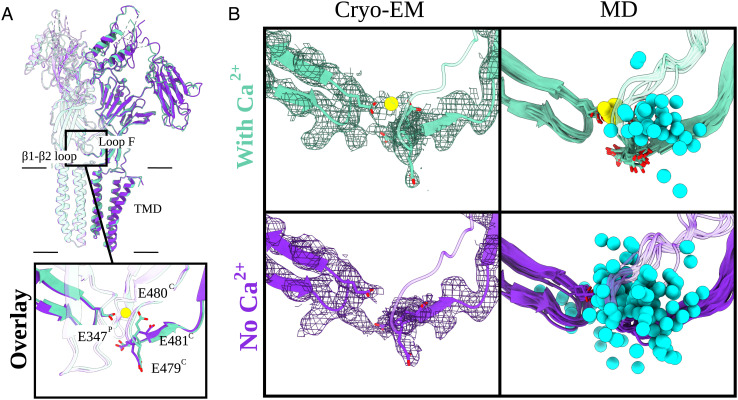
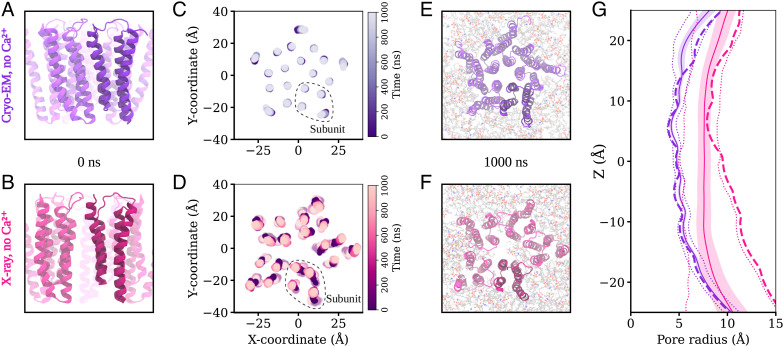
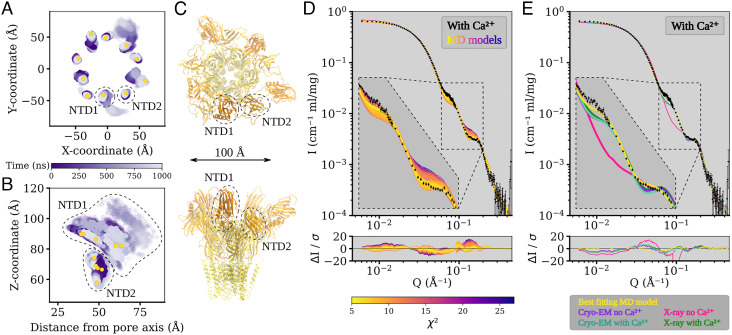
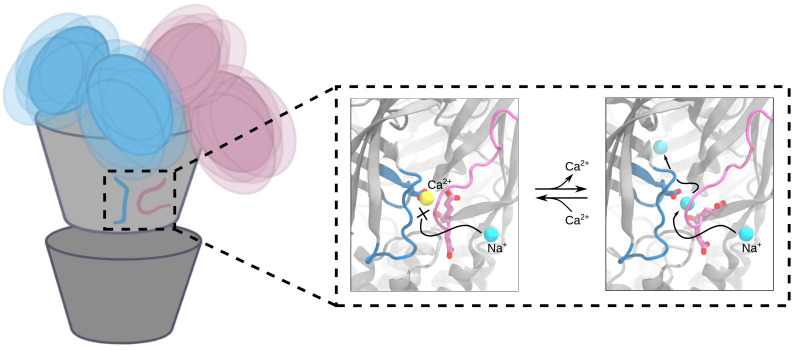
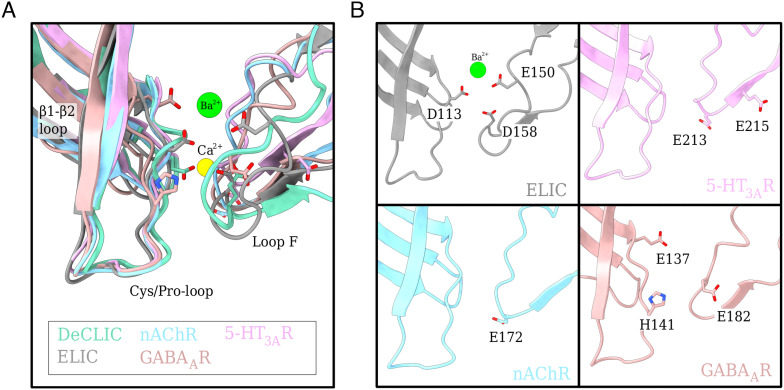
Pentameric ligand-gated ion channels (pLGICs) perform electrochemical signal transduction in organisms ranging from bacteria to humans. Among the prokaryotic pLGICs, there is architectural diversity involving N-terminal domains (NTDs) not found in eukaryotic relatives, exemplified by the calcium-sensitive channel (DeCLIC) from a Desulfofustis deltaproteobacterium, which has an NTD in addition to the canonical pLGIC structure. Here, we have characterized the structure and dynamics of DeCLIC through cryoelectron microscopy (cryo-EM), small-angle neutron scattering (SANS), and molecular dynamics (MD) simulations. In the presence and absence of calcium, cryo-EM yielded structures with alternative conformations of the calcium-binding site. SANS profiles further revealed conformational diversity at room temperature beyond that observed in static structures, shown through MD to be largely attributable to rigid-body motions of the NTD relative to the protein core, with expanded and asymmetric conformations improving the fit of the SANS data. This work reveals the range of motion available to the DeCLIC NTD and calcium-binding site, expanding the conformational landscape of the pLGIC family. Further, these findings demonstrate the power of combining low-resolution scattering, high-resolution structural, and MD simulation data to elucidate interfacial interactions that are highly conserved in the pLGIC family.
Keywords: Cys-loop receptors; calcium; ligand-gated ion channel; small-angle neutron scattering.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Nemecz Á., Prevost M. S., Menny A., Corringer P. J., Emerging molecular mechanisms of signal transduction in pentameric ligand-gated ion channels. Neuron 90, 452–470 (2016). - PubMed
-
- Sauguet L., Shahsavar A., Delarue M., Crystallographic studies of pharmacological sites in pentameric ligand-gated ion channels. Biochim. Biophysi. Acta (BBA)-Gen. Subj. 1850, 511–523 (2015). - PubMed
-
- Taly A., Corringer P. J., Guedin D., Lestage P., Changeux J. P., Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Dis. 8, 733–750 (2009). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
