

Non-genetic adaptive resistance to KRASG12C inhibition: EMT is not the only culprit
- PMID: 36483040
- PMCID: PMC9722758
- DOI: 10.3389/fonc.2022.1004669
Non-genetic adaptive resistance to KRASG12C inhibition: EMT is not the only culprit
Abstract
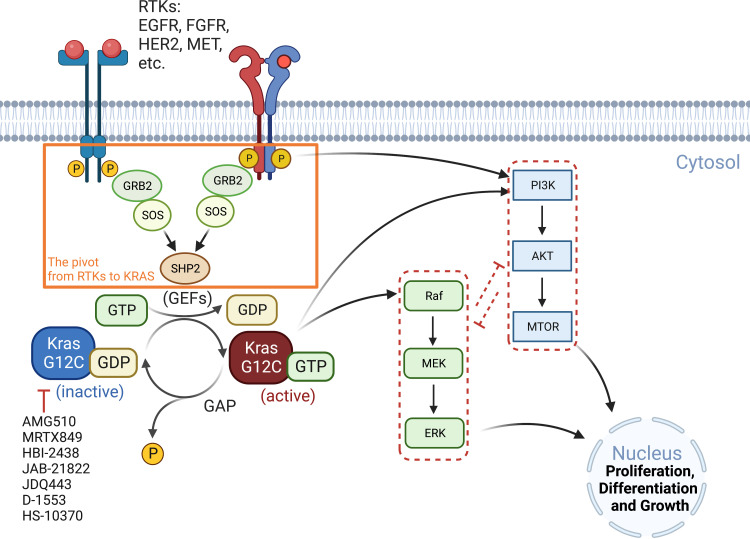
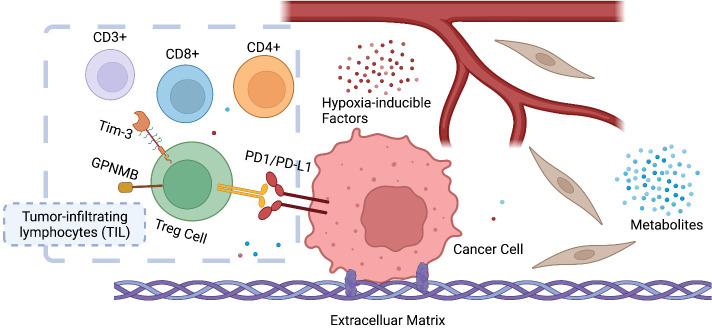
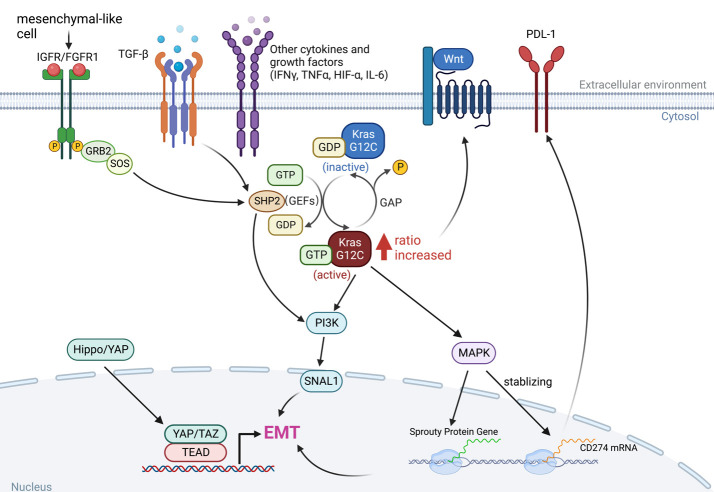
Adaptions to therapeutic pressures exerted on cancer cells enable malignant progression of the tumor, culminating in escape from programmed cell death and development of resistant diseases. A common form of cancer adaptation is non-genetic alterations that exploit mechanisms already present in cancer cells and do not require genetic modifications that can also lead to resistance mechanisms. Epithelial-to-mesenchymal transition (EMT) is one of the most prevalent mechanisms of adaptive drug resistance and resulting cancer treatment failure, driven by epigenetic reprogramming and EMT-specific transcription factors. A recent breakthrough in cancer treatment is the development of KRASG12C inhibitors, which herald a new era of therapy by knocking out a unique substitution of an oncogenic driver. However, these highly selective agents targeting KRASG12C, such as FDA-approved sotorasib (AMG510) and adagrasib (MRTX849), inevitably encounter multiple mechanisms of drug resistance. In addition to EMT, cancer cells can hijack or rewire the sophisticated signaling networks that physiologically control cell proliferation, growth, and differentiation to promote malignant cancer cell phenotypes, suggesting that inhibition of multiple interconnected signaling pathways may be required to block tumor progression on KRASG12C inhibitor therapy. Furthermore, the tumor microenvironment (TME) of cancer cells, such as tumor-infiltrating lymphocytes (TILs), contribute significantly to immune escape and tumor progression, suggesting a therapeutic approach that targets not only cancer cells but also the TME. Deciphering and targeting cancer adaptions promises mechanistic insights into tumor pathobiology and improved clinical management of KRASG12C-mutant cancer. This review presents recent advances in non-genetic adaptations leading to resistance to KRASG12C inhibitors, with a focus on oncogenic pathway rewiring, TME, and EMT.
Keywords: EMT; KRAS G12C inhibitors; TME; non-genetic adaptive resistance; symbiosis.
Copyright © 2022 Ning, Marti, Dorn and Peng.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Dual inhibition of HERs and PD-1 counteract resistance in KRASG12C-mutant head and neck cancer.J Exp Clin Cancer Res. 2024 Nov 20;43(1):308. doi: 10.1186/s13046-024-03227-0. J Exp Clin Cancer Res. 2024. PMID: 39567998 Free PMC article.
-
A Breakthrough Brought about by Targeting KRASG12C: Nonconformity Is Punished.Cancers (Basel). 2022 Jan 13;14(2):390. doi: 10.3390/cancers14020390. Cancers (Basel). 2022. PMID: 35053550 Free PMC article. Review.
-
Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant Non-Small Cell Lung Cancer.Clin Cancer Res. 2020 Nov 15;26(22):5962-5973. doi: 10.1158/1078-0432.CCR-20-2077. Epub 2020 Sep 8. Clin Cancer Res. 2020. PMID: 32900796
-
Assessment of KRASG12C inhibitors for colorectal cancer.Front Oncol. 2024 Jun 24;14:1412435. doi: 10.3389/fonc.2024.1412435. eCollection 2024. Front Oncol. 2024. PMID: 38978742 Free PMC article. Review.
-
Resistance to KRASG12C Inhibitors in Non-Small Cell Lung Cancer.Front Oncol. 2021 Dec 24;11:787585. doi: 10.3389/fonc.2021.787585. eCollection 2021. Front Oncol. 2021. PMID: 35004309 Free PMC article. Review.
Cited by
-
Sotorasib resistance triggers epithelial-mesenchymal transition and activates AKT and P38-mediated signaling.Front Mol Biosci. 2025 Jan 30;12:1537523. doi: 10.3389/fmolb.2025.1537523. eCollection 2025. Front Mol Biosci. 2025. PMID: 39950162 Free PMC article.
-
The Extracellular Niche and Tumor Microenvironment Enhance KRAS Inhibitor Efficacy in Pancreatic Cancer.Cancer Res. 2024 Apr 1;84(7):1115-1132. doi: 10.1158/0008-5472.CAN-23-2504. Cancer Res. 2024. PMID: 38294344 Free PMC article.
-
Targeting KRAS in Colorectal Cancer: A Bench to Bedside Review.Int J Mol Sci. 2023 Jul 27;24(15):12030. doi: 10.3390/ijms241512030. Int J Mol Sci. 2023. PMID: 37569406 Free PMC article. Review.
-
FBXL16 promotes cell growth and drug resistance in lung adenocarcinomas with KRAS mutation by stabilizing IRS1 and upregulating IRS1/AKT signaling.Mol Oncol. 2024 Mar;18(3):762-777. doi: 10.1002/1878-0261.13554. Epub 2024 Jan 17. Mol Oncol. 2024. PMID: 37983945 Free PMC article.
-
KRAS: Biology, Inhibition, and Mechanisms of Inhibitor Resistance.Curr Oncol. 2024 Apr 3;31(4):2024-2046. doi: 10.3390/curroncol31040150. Curr Oncol. 2024. PMID: 38668053 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous