

Long-term health outcomes following curative therapies for sickle cell disease
- PMID: 36485115
- PMCID: PMC9820909
- DOI: 10.1182/hematology.2022000373
Long-term health outcomes following curative therapies for sickle cell disease
Abstract
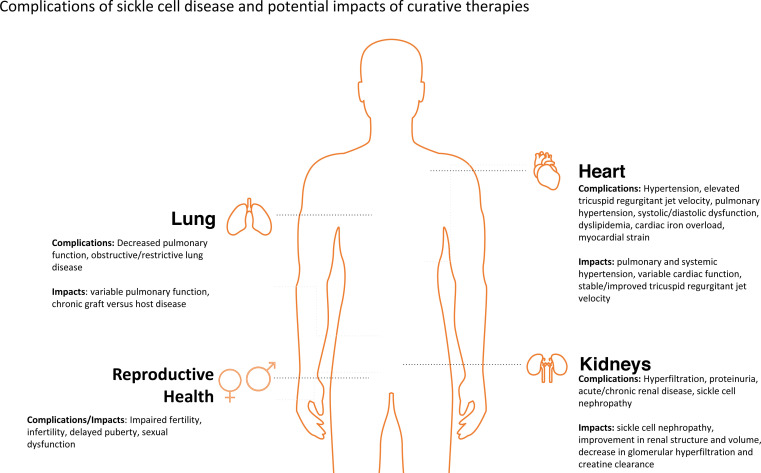
Treatment options for patients with sickle cell disease (SCD) continue to rapidly expand and evolve. The goal of therapies such as an allogeneic hematopoietic stem cell transplant (HSCT), gene therapy, and gene editing is to cure rather than control SCD. The benefits of these therapies must be accompanied by minimizing long-term adverse health outcomes from SCD and its treatment. SCD can have adverse effects on a variety of organ systems, including the heart, lung, kidney, and reproductive system, leading to high disease burden, morbidity, and premature mortality in both pediatric and adult patients. While curative therapies are being increasingly used, there remains a paucity of data on the long-term health outcomes associated with these treatments in children and adults with SCD. There are data available regarding the effects of HSCT performed largely for malignant diseases, from which data on SCD outcomes may be extrapolated. However, given the significant differences between these 2 populations of patients who undergo HSCT, such extrapolation is imprecise at best. Furthermore, there are currently no published data on long-term health outcomes following gene therapy for SCD due to current short follow-up times. We summarize the limited data reported on health outcomes following HSCT for SCD and emphasize the need for more research within this area.
Copyright © 2022 by The American Society of Hematology.
Conflict of interest statement
Rohini Chakravarthy: no competing financial interests to declare.
Debra L. Friedman: no competing financial interests to declare.
Figures


References
-
- Osunkwo I, Andemariam B, Minniti CP, et al.. Impact of sickle cell disease on patients' daily lives, symptoms reported, and disease management strategies: results from the international Sickle Cell World Assessment Survey (SWAY). Am J Hematol. 2021;96(4):404-417. doi:10.1002/ajh.26063. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical