

High-throughput robust single-cell DNA methylation profiling with sciMETv2
- PMID: 36494343
- PMCID: PMC9734657
- DOI: 10.1038/s41467-022-35374-3
High-throughput robust single-cell DNA methylation profiling with sciMETv2
Abstract
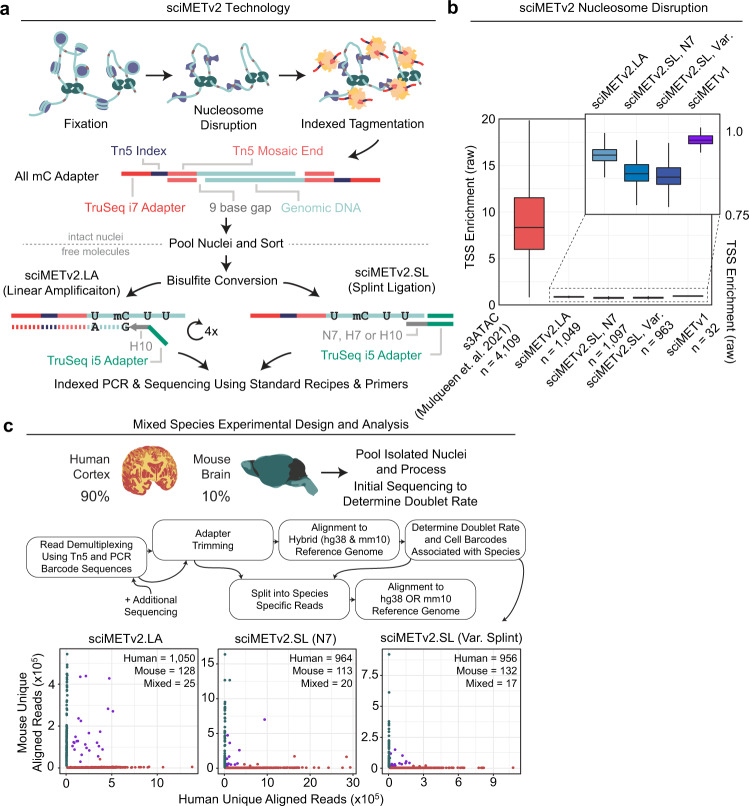
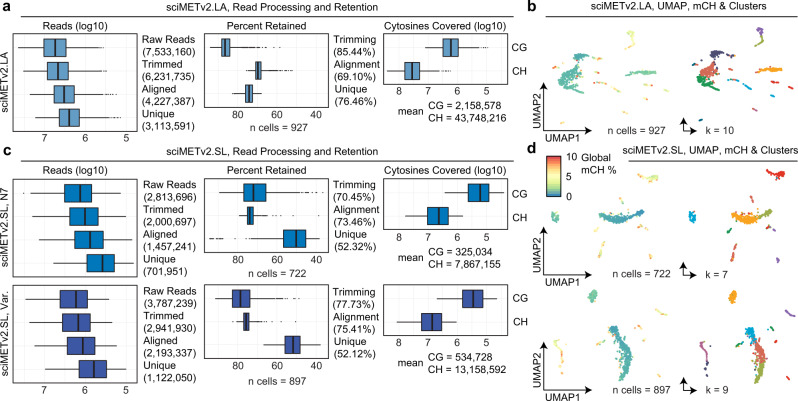
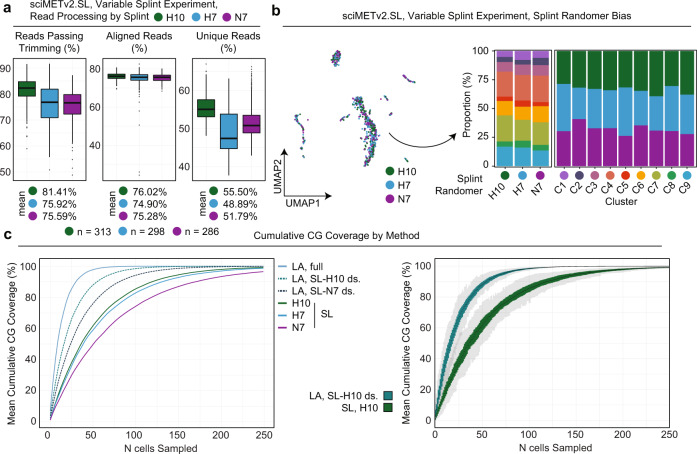
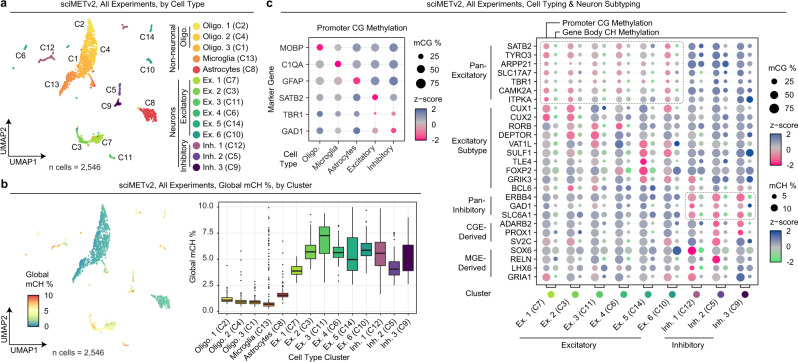
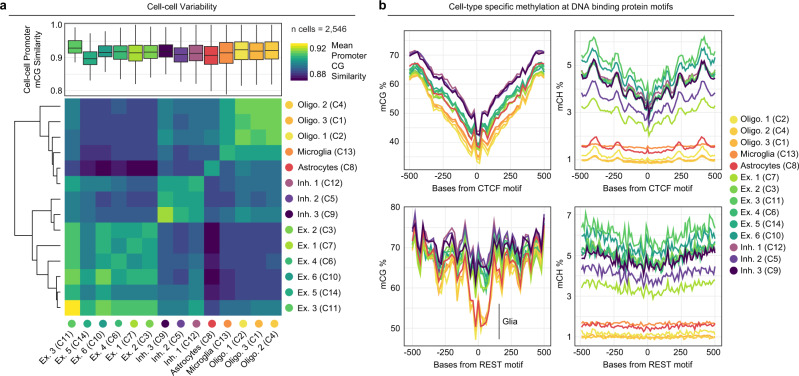
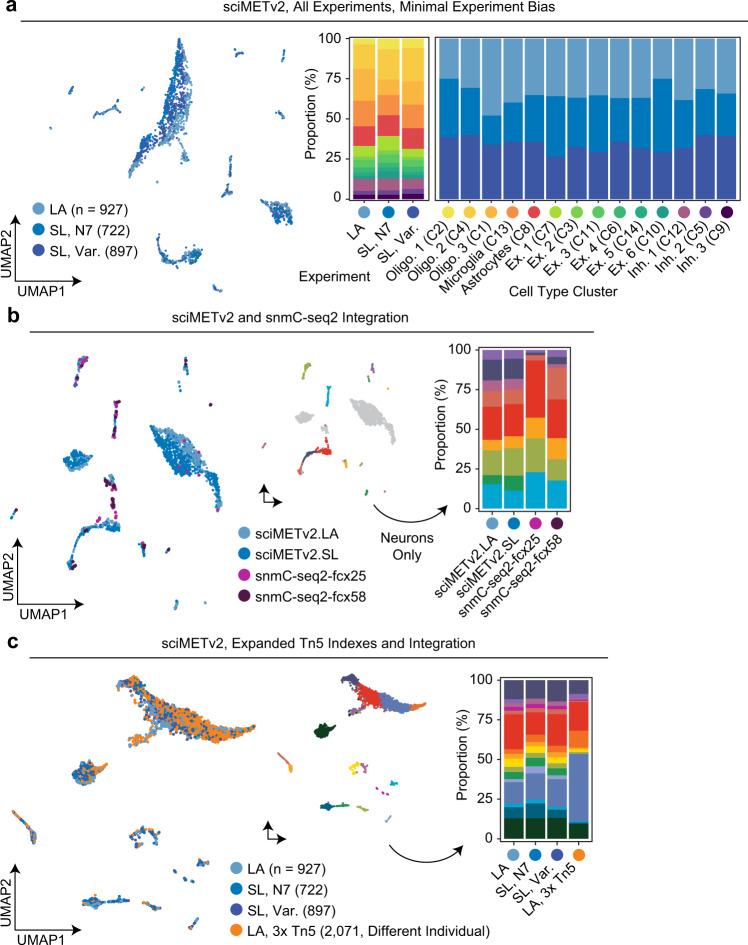
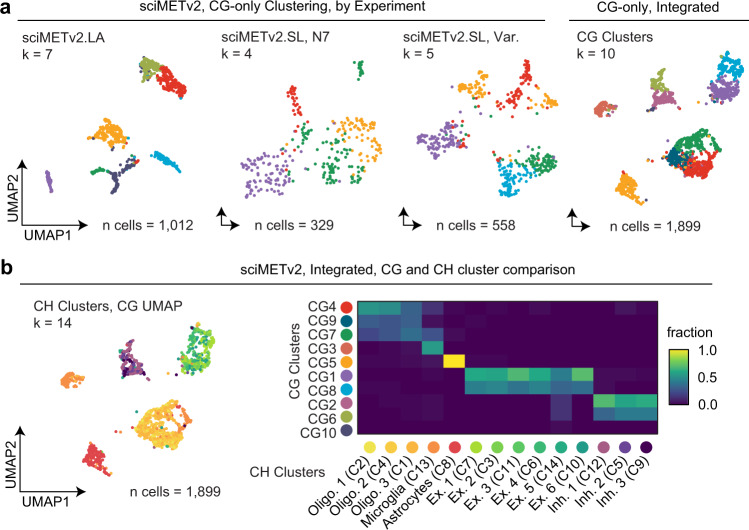
DNA methylation is a key epigenetic property that drives gene regulatory programs in development and disease. Current single-cell methods that produce high quality methylomes are expensive and low throughput without the aid of extensive automation. We previously described a proof-of-principle technique that enabled high cell throughput; however, it produced only low-coverage profiles and was a difficult protocol that required custom sequencing primers and recipes and frequently produced libraries with excessive adapter contamination. Here, we describe a greatly improved version that generates high-coverage profiles (~15-fold increase) using a robust protocol that does not require custom sequencing capabilities, includes multiple stopping points, and exhibits minimal adapter contamination. We demonstrate two versions of sciMETv2 on primary human cortex, a high coverage and rapid version, identifying distinct cell types using CH methylation patterns. These datasets are able to be directly integrated with one another as well as with existing snmC-seq2 datasets with little discernible bias. Finally, we demonstrate the ability to determine cell types using CG methylation alone, which is the dominant context for DNA methylation in most cell types other than neurons and the most applicable analysis outside of brain tissue.
© 2022. The Author(s).
Conflict of interest statement
J.T., D.P., and F.J.S. are employees of Scale Biosciences. R.M.M., F.J.S., and A.C.A. are authors on licensed patents that cover the nucleosome disruption and indexed tagmentation design components of the technologies described in this manuscript (WO2018018008A1 and WO2018226708A1, granted). This potential conflict of interest for A.C.A. and R.M.M. has been reviewed and managed by OHSU. The remaining authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
