

Clinical and genetic characteristics of Chinese patients with congenital cranial dysinnervation disorders
- PMID: 36494820
- PMCID: PMC9733177
- DOI: 10.1186/s13023-022-02582-5
Clinical and genetic characteristics of Chinese patients with congenital cranial dysinnervation disorders
Abstract
Background: Congenital cranial dysinnervation disorders (CCDDs) are a group of diseases with high clinical and genetic heterogeneity. Clinical examinations combined with Magnetic resonance imaging (MRI) and whole exome sequencing (WES) were performed to reveal the phenotypic and genotypic characteristics in a cohort of Chinese CCDDs patients.
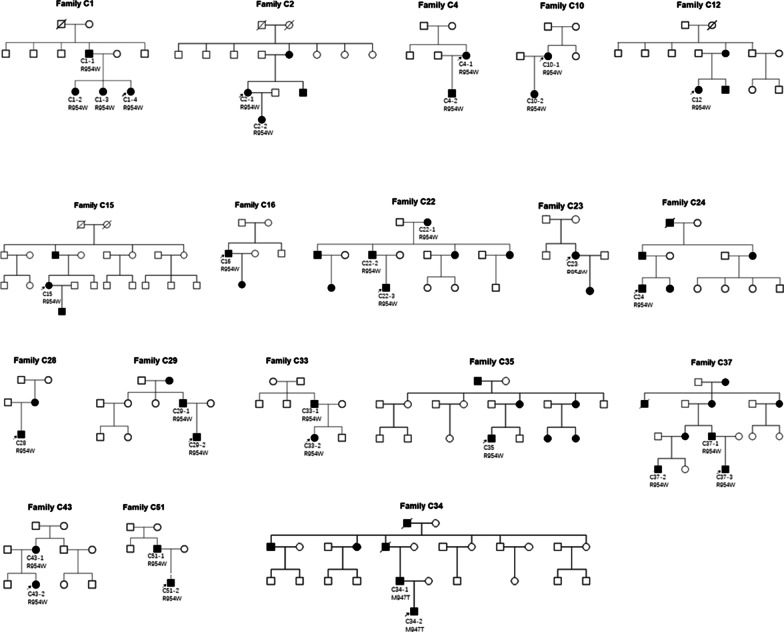
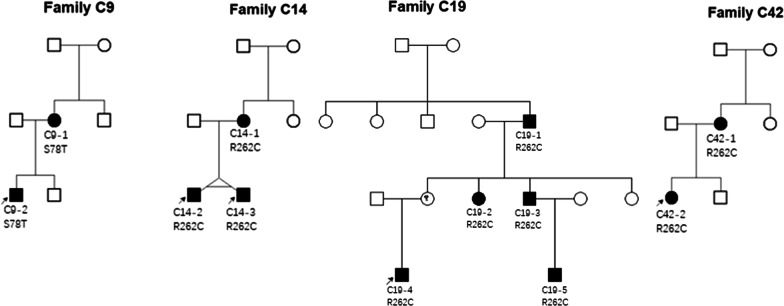
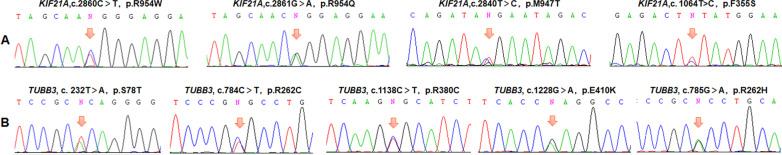
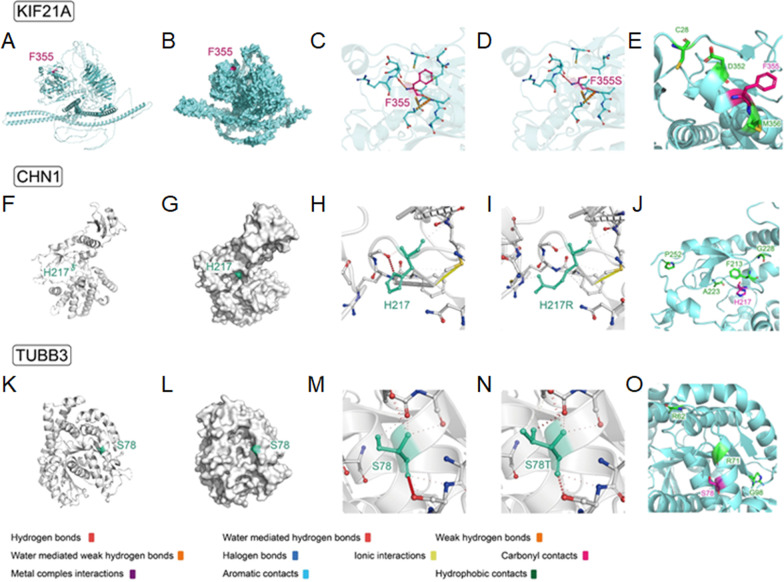
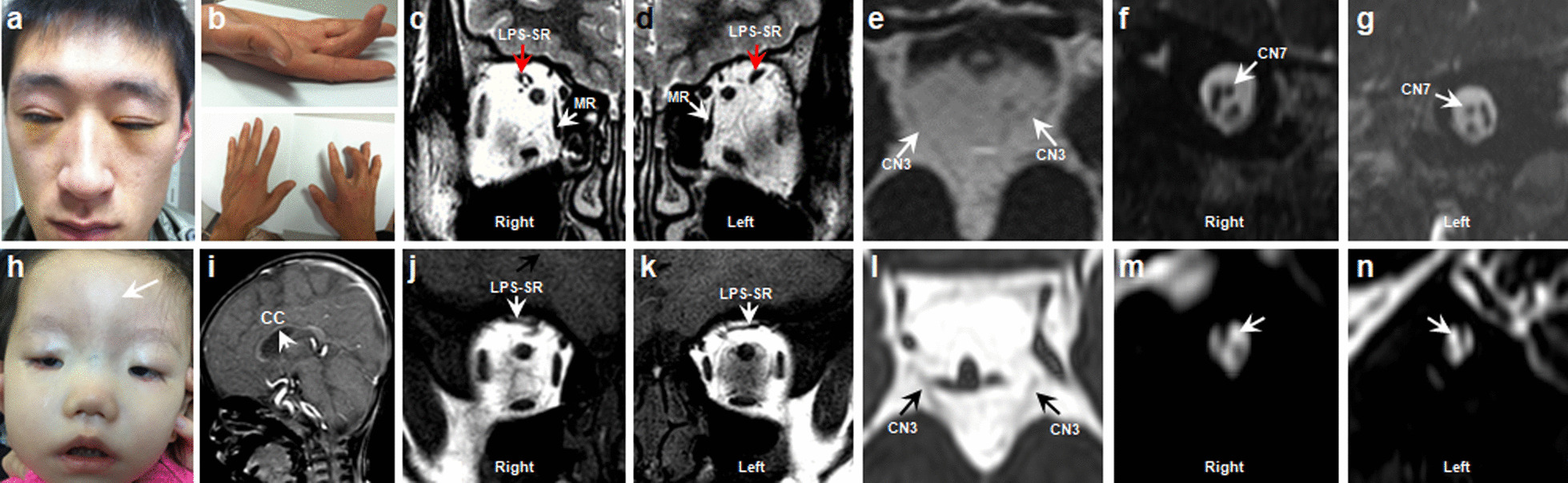
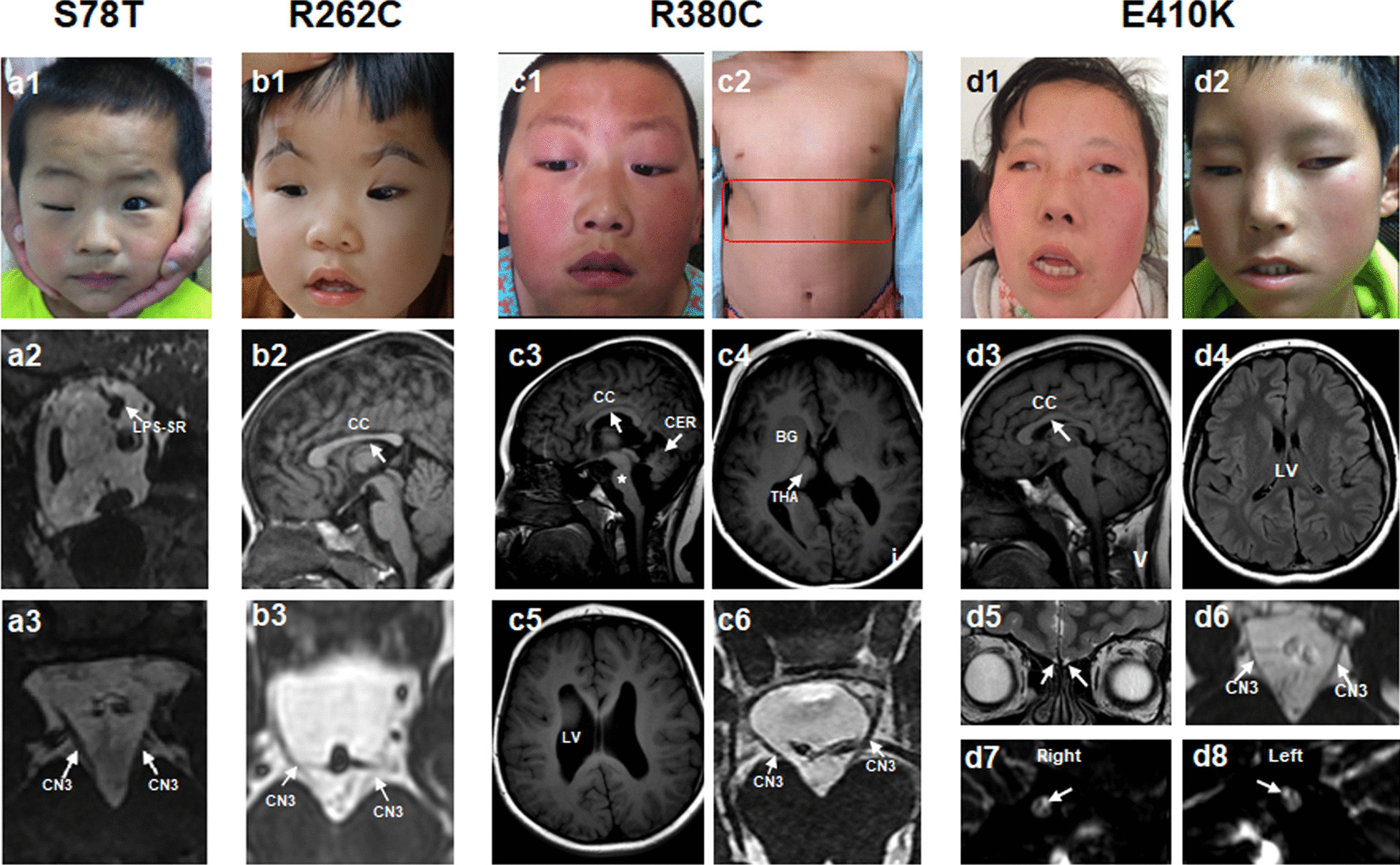
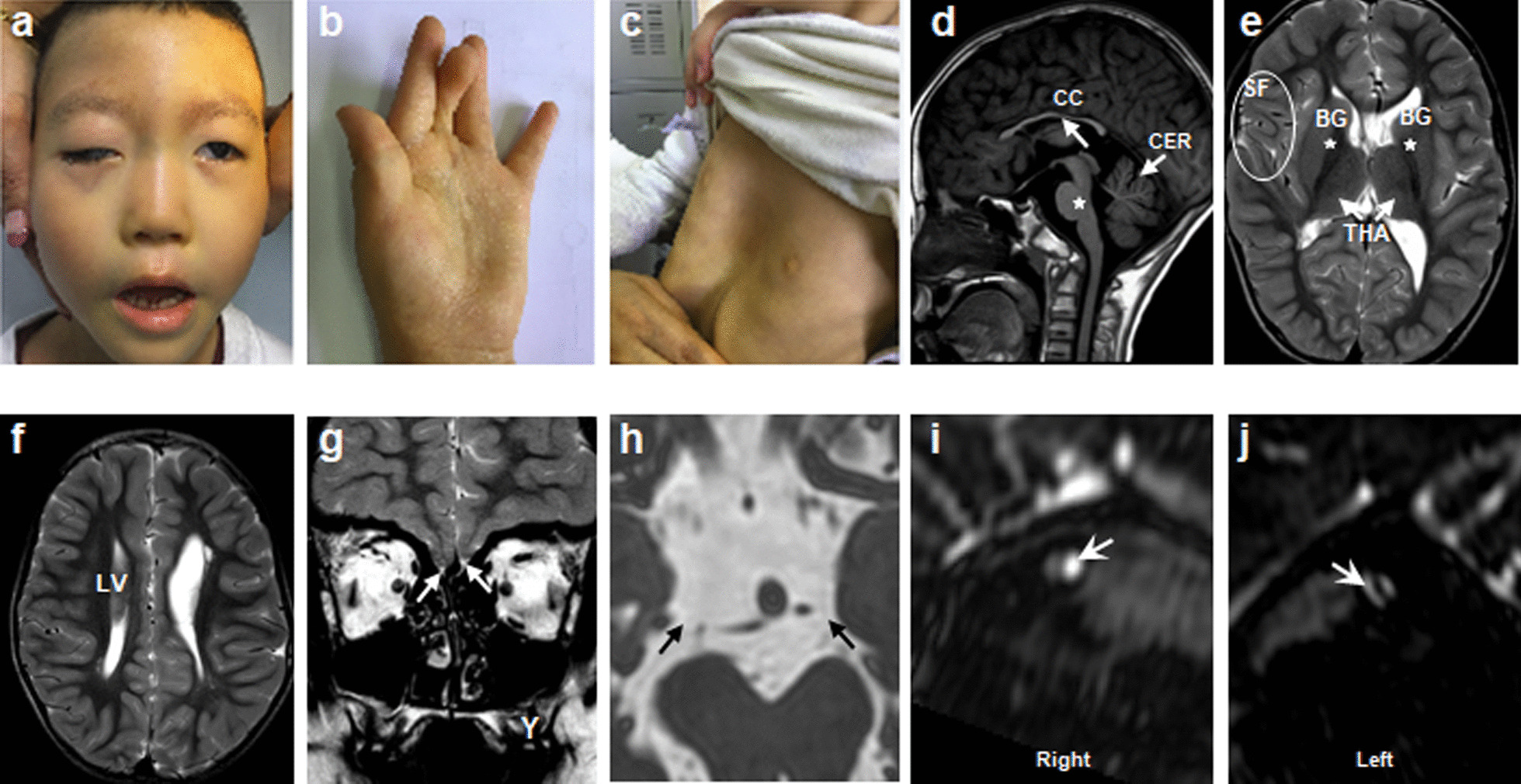
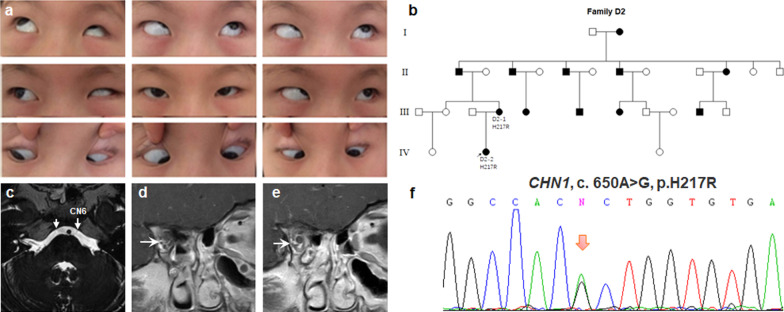
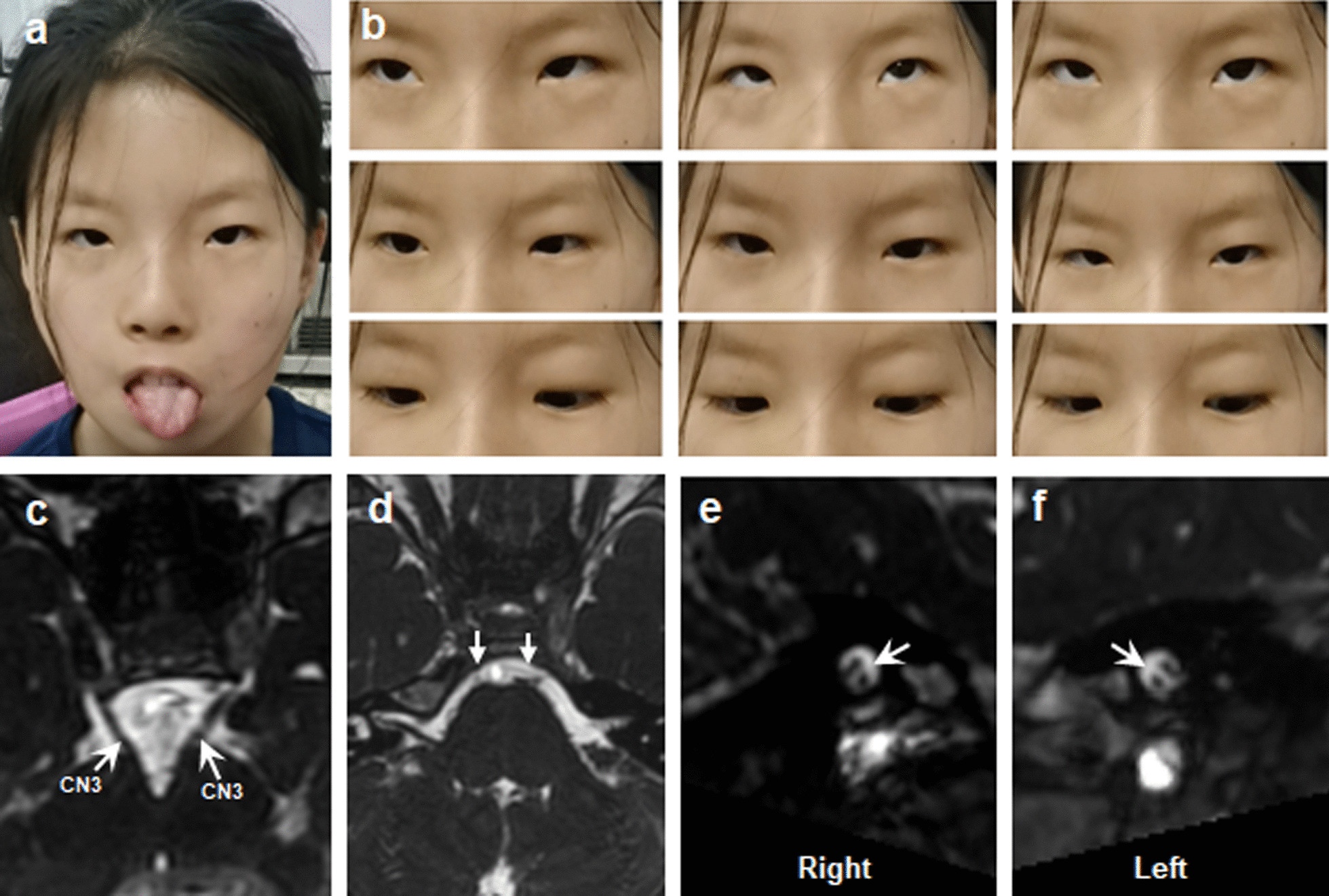
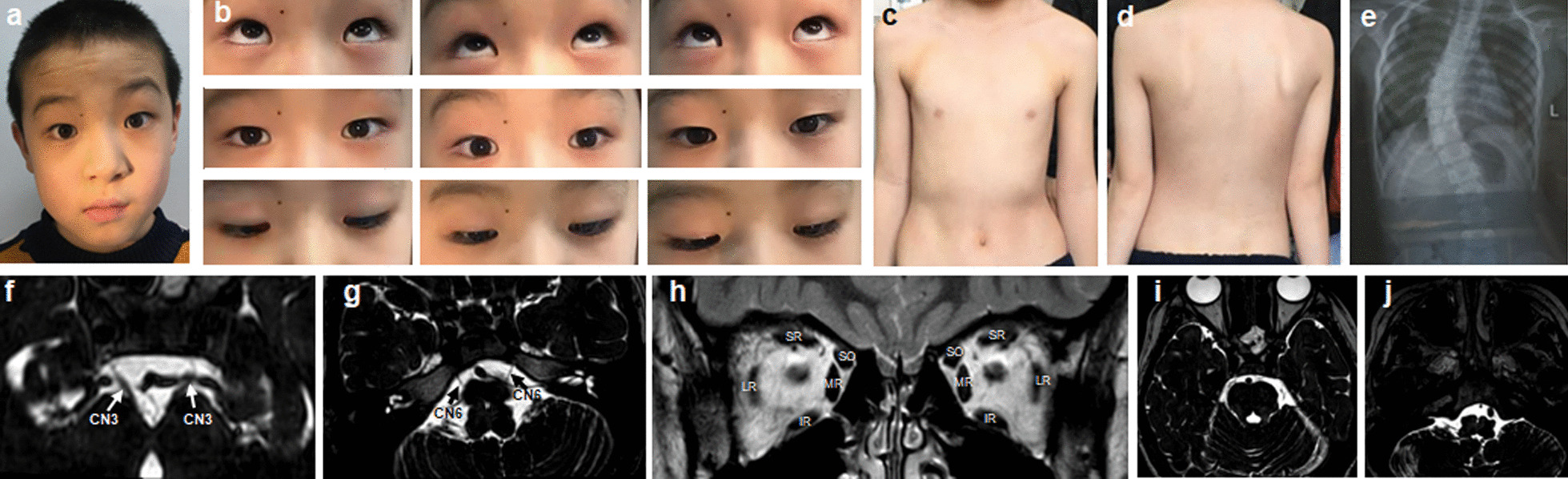
Results: A total of 122 CCDDs patients from 96 families were enrolled. All patients showed restrictive eye movements, and 46 patients from 46 families (47.9%, 46/96) were accompanied by multiple congenital malformations. Multi-positional high-resolution MRI was performed in 94 patients from 88 families, of which, all patients had hypoplasia of the cranial nerves except HGPPS patients and 15 patients from 15 families (17.0%,15/88) were accompanied by other craniocerebral malformations. WES was performed in 122 CCDDs patients. Ten pathogenic variants were detected in KIF21A, TUBB3, and CHN1 genes in 43 families. Three variants were unreported, including KIF21A (c.1064T > C, p.F355S), TUBB3 (c.232T > A, p.S78T) and CHN1 (c.650A > G, p.H217R). Of the 43 probands harboring pathogenic variants, 42 were diagnosed with Congenital Fibrosis of Extraocular Muscles (CFEOM) and one was Duane Retraction Syndrome (DRS). No definite pathogenic variants in known candidate genes of CCDDs were found in sporadic DRS, Möbius Syndrome (MBS) and Horizontal Gaze Palsy with Progressive Scoliosis (HGPPS) patients. The CFEOM patients harboring R380C, E410K and R262H variants in TUBB3 gene and F355S variant in KIF21A gene exhibited syndromic phenotypes.
Conclusions: This study broadened the phenotypic and genotypic spectrums of CCDDs, and it was the largest clinical and genetic investigation for CCDDs patients from China. KIF21A and TUBB3 were the common pathogenic genes in Chinese CFEOM. MRI coupled with WES can provide a supportive diagnosis in patients with clinically suspected CCDDs.
Keywords: Congenital cranial dysinnervation disorders; Genetics; Magnetic resonance imaging; Phenotypic and genotypic characteristics; Whole exome sequencing.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Gutowski NJ, Bosley TM, Engle EC. 110th ENMC International Workshop: the congenital cranial dysinnervation disorders (CCDDs). Naarden, The Netherlands, 25–27 October, 2002. Neuromuscul Disord 2003;13(7–8):573–8. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
