

Spatial-ID: a cell typing method for spatially resolved transcriptomics via transfer learning and spatial embedding
- PMID: 36496406
- PMCID: PMC9741613
- DOI: 10.1038/s41467-022-35288-0
Spatial-ID: a cell typing method for spatially resolved transcriptomics via transfer learning and spatial embedding
Abstract
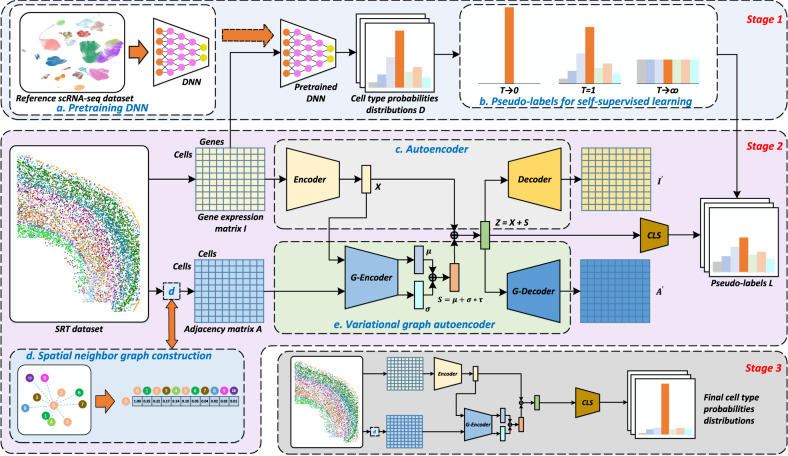
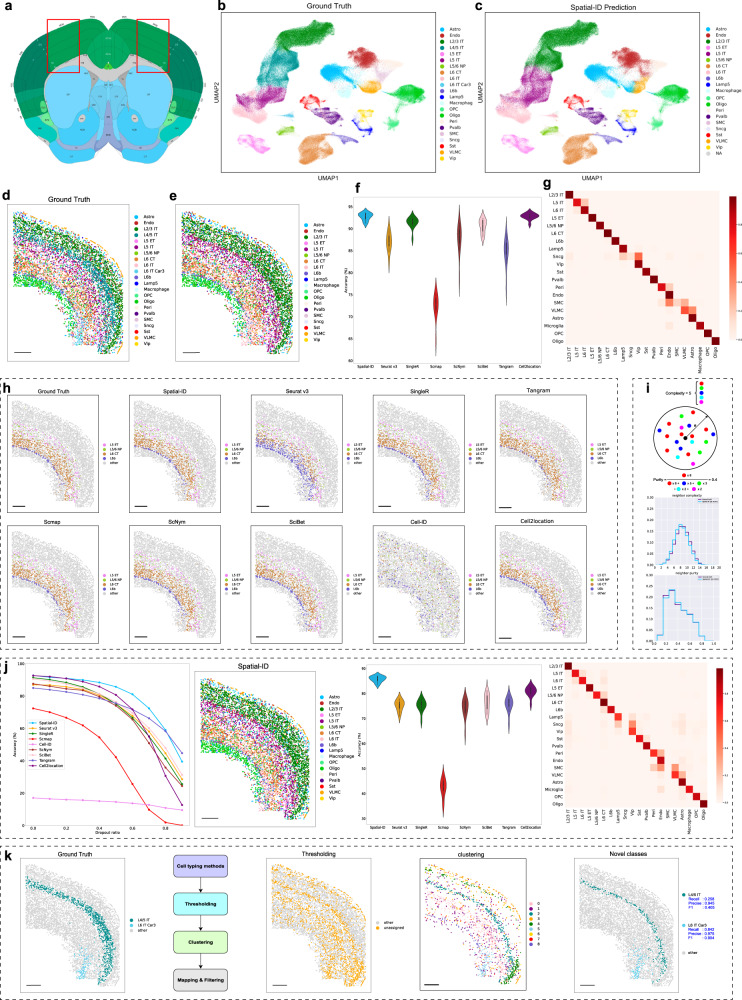
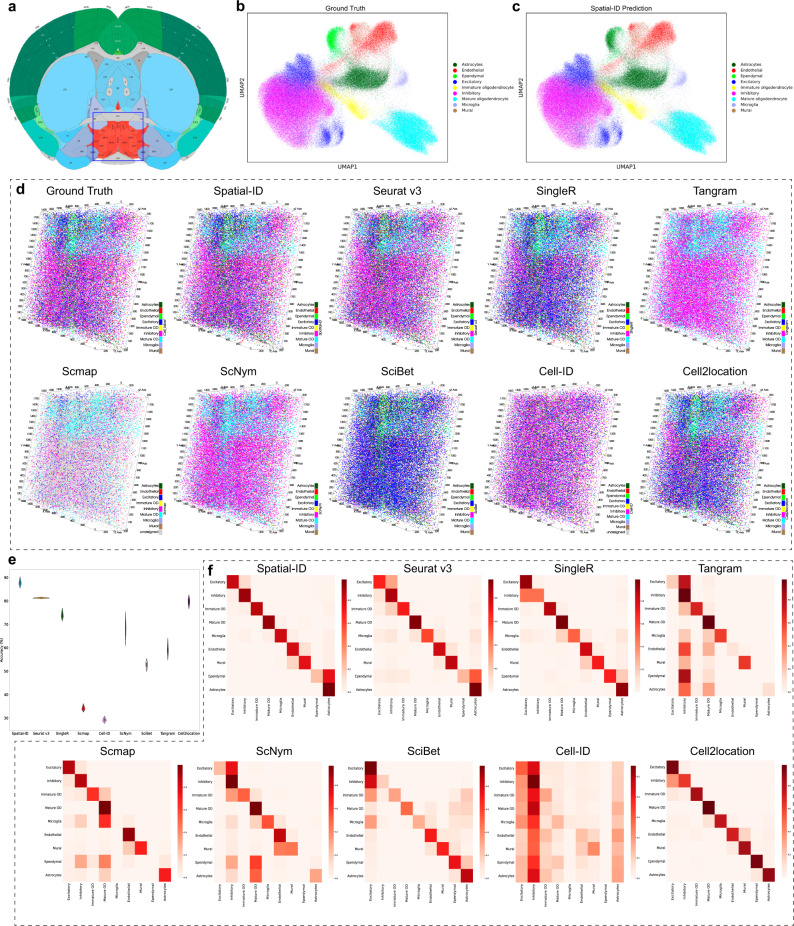
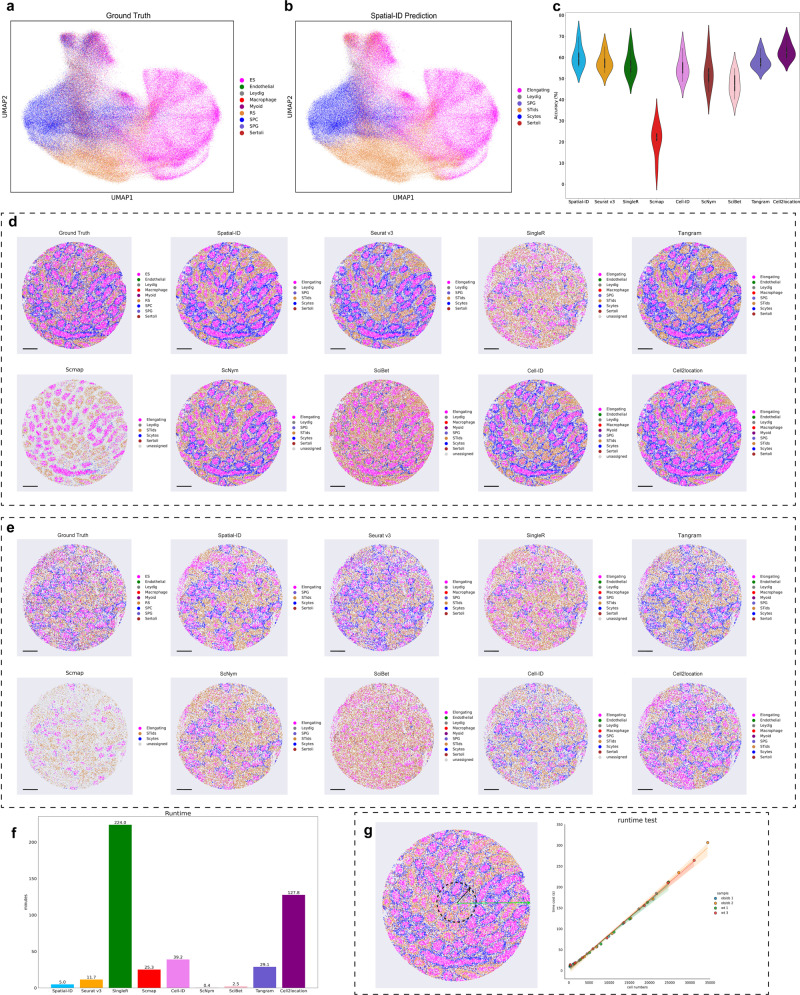
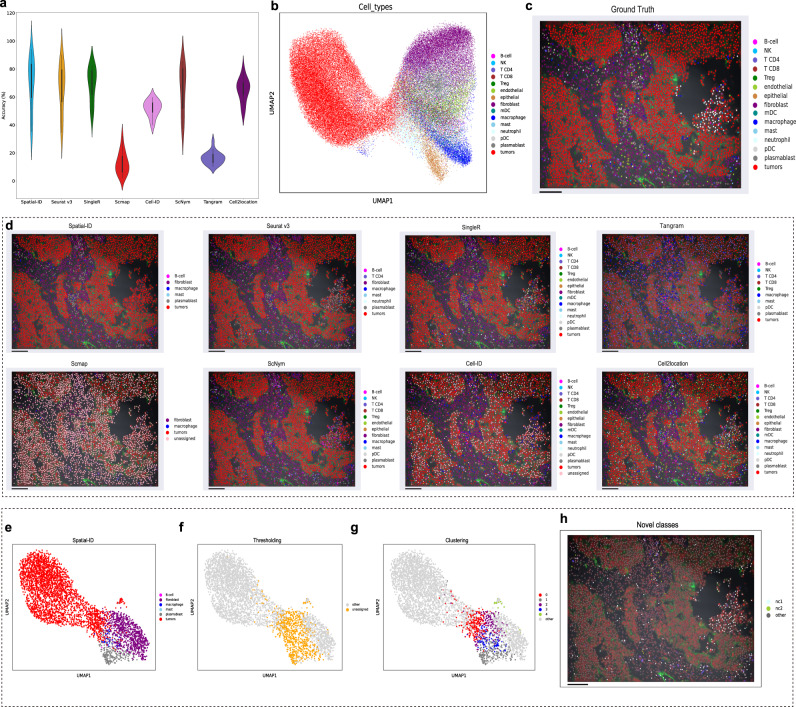
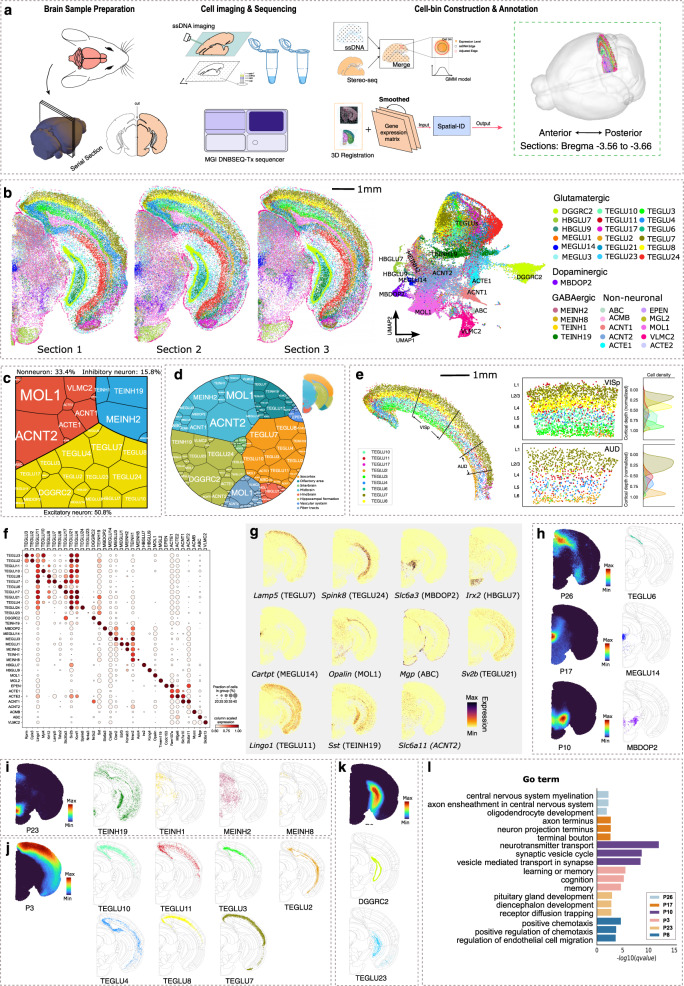
Spatially resolved transcriptomics provides the opportunity to investigate the gene expression profiles and the spatial context of cells in naive state, but at low transcript detection sensitivity or with limited gene throughput. Comprehensive annotating of cell types in spatially resolved transcriptomics to understand biological processes at the single cell level remains challenging. Here we propose Spatial-ID, a supervision-based cell typing method, that combines the existing knowledge of reference single-cell RNA-seq data and the spatial information of spatially resolved transcriptomics data. We present a series of benchmarking analyses on publicly available spatially resolved transcriptomics datasets, that demonstrate the superiority of Spatial-ID compared with state-of-the-art methods. Besides, we apply Spatial-ID on a self-collected mouse brain hemisphere dataset measured by Stereo-seq, that shows the scalability of Spatial-ID to three-dimensional large field tissues with subcellular spatial resolution.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Graph domain adaptation-based framework for gene expression enhancement and cell type identification in large-scale spatially resolved transcriptomics.Brief Bioinform. 2024 Sep 23;25(6):bbae576. doi: 10.1093/bib/bbae576. Brief Bioinform. 2024. PMID: 39508445 Free PMC article.
-
scBOL: a universal cell type identification framework for single-cell and spatial transcriptomics data.Brief Bioinform. 2024 Mar 27;25(3):bbae188. doi: 10.1093/bib/bbae188. Brief Bioinform. 2024. PMID: 38678389 Free PMC article.
-
Probabilistic embedding, clustering, and alignment for integrating spatial transcriptomics data with PRECAST.Nat Commun. 2023 Jan 18;14(1):296. doi: 10.1038/s41467-023-35947-w. Nat Commun. 2023. PMID: 36653349 Free PMC article.
-
Computational solutions for spatial transcriptomics.Comput Struct Biotechnol J. 2022 Sep 1;20:4870-4884. doi: 10.1016/j.csbj.2022.08.043. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36147664 Free PMC article. Review.
-
Challenges and considerations for single-cell and spatially resolved transcriptomics sample collection during spaceflight.Cell Rep Methods. 2022 Oct 31;2(11):100325. doi: 10.1016/j.crmeth.2022.100325. eCollection 2022 Nov 21. Cell Rep Methods. 2022. PMID: 36452864 Free PMC article. Review.
Cited by
-
Graph domain adaptation-based framework for gene expression enhancement and cell type identification in large-scale spatially resolved transcriptomics.Brief Bioinform. 2024 Sep 23;25(6):bbae576. doi: 10.1093/bib/bbae576. Brief Bioinform. 2024. PMID: 39508445 Free PMC article.
-
Exploit Spatially Resolved Transcriptomic Data to Infer Cellular Features from Pathology Imaging Data.bioRxiv [Preprint]. 2024 Aug 7:2024.08.05.606654. doi: 10.1101/2024.08.05.606654. bioRxiv. 2024. PMID: 39149252 Free PMC article. Preprint.
-
Spotiphy enables single-cell spatial whole transcriptomics across an entire section.Nat Methods. 2025 Apr;22(4):724-736. doi: 10.1038/s41592-025-02622-5. Epub 2025 Mar 12. Nat Methods. 2025. PMID: 40074951 Free PMC article.
-
Detecting anomalous anatomic regions in spatial transcriptomics with STANDS.Nat Commun. 2024 Sep 19;15(1):8223. doi: 10.1038/s41467-024-52445-9. Nat Commun. 2024. PMID: 39300113 Free PMC article.
-
Spatiotemporal transcriptomic atlas of rhizome formation in Oryza longistaminata.Plant Biotechnol J. 2024 Jun;22(6):1652-1668. doi: 10.1111/pbi.14294. Epub 2024 Feb 12. Plant Biotechnol J. 2024. PMID: 38345936 Free PMC article.
References
MeSH terms
LinkOut - more resources
Full Text Sources
