

Systematic Review of Clinical and Pathophysiological Features of Genetic Creutzfeldt-Jakob Disease Caused by a Val-to-Ile Mutation at Codon 180 in the Prion Protein Gene
- PMID: 36499498
- PMCID: PMC9737045
- DOI: 10.3390/ijms232315172
Systematic Review of Clinical and Pathophysiological Features of Genetic Creutzfeldt-Jakob Disease Caused by a Val-to-Ile Mutation at Codon 180 in the Prion Protein Gene
Abstract
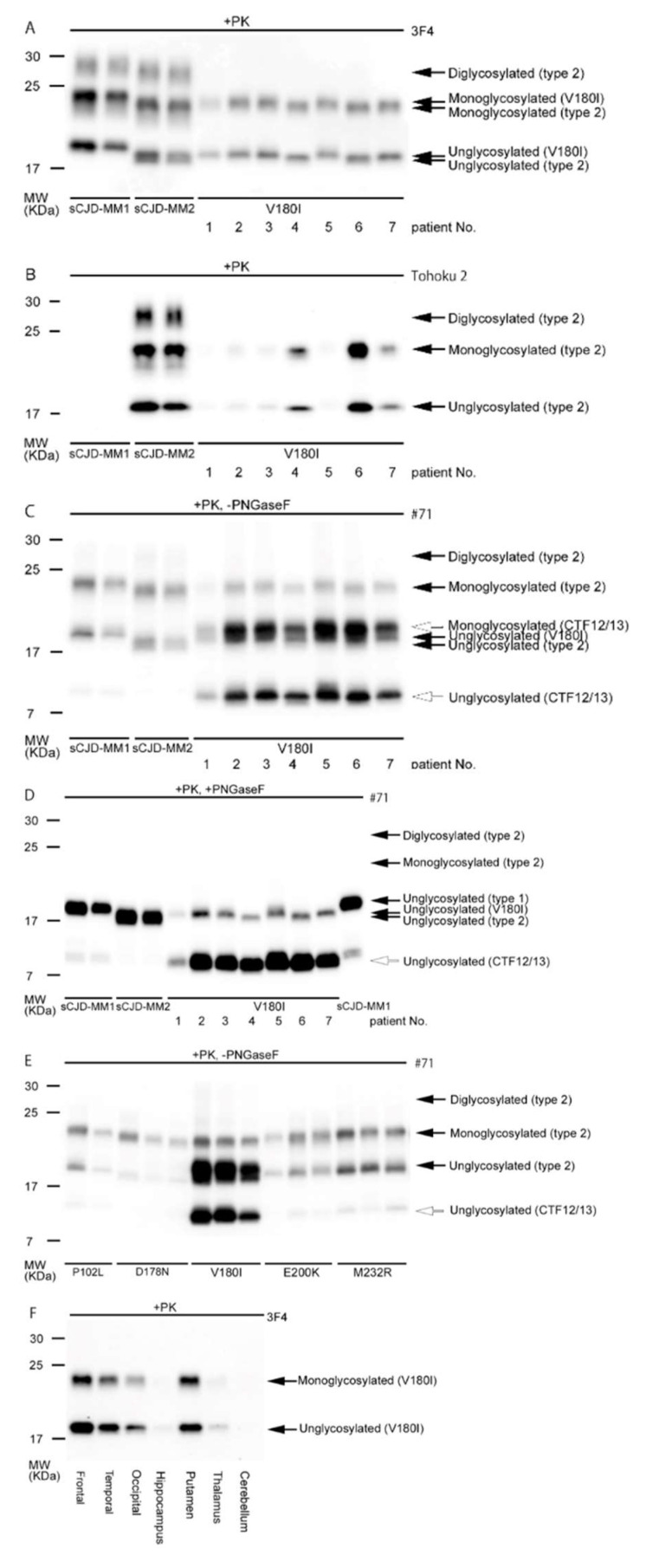
Genetic Creutzfeldt-Jakob disease (gCJD) is a subtype of genetic prion diseases (gPrDs) caused by the accumulation of mutated pathological prion proteins (PrPSc). gCJD has a phenotypic similarity with sporadic CJD (sCJD). In Japan, gCJD with a Val to Ile substitution at codon 180 (V180I-gCJD) is the most frequent gPrD, while the mutation is extremely rare in countries other than Japan and Korea. In this article, we aim to review previously elucidated clinical and biochemical features of V180I-gCJD, expecting to advance the understanding of this unique subtype in gCJD. Compared to classical sCJD, specific clinical features of V180I-gCJD include older age at onset, a relatively slow progression of dementia, and a lower positivity for developing myoclonus, cerebellar, pyramidal signs, and visual disturbance. Diffuse edematous ribboning hyperintensity of the cerebral cortex, without occipital lobes in diffusion-weighted magnetic resonance imaging, is also specific. Laboratory data reveal the low positivity of PrPSc in the cerebrospinal fluid and periodic sharp wave complexes on an electroencephalogram. Most patients with V180I-gCJD have been reported to have no family history, probably due to the older age at onset, and clinical and biochemical features indicate the specific phenotype associated with the prion protein gene mutation.
Keywords: V180I; Val-to-Ile substitution at codon 180; genetic Creutzfeldt–Jakob disease; genetic prion disease; normal prion proteins; pathological prion proteins; prion protein gene.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
Characterization of prion strains and peripheral prion infectivity patterns in E200K genetic CJD patients.Acta Neuropathol. 2025 Jun 16;149(1):62. doi: 10.1007/s00401-025-02903-5. Acta Neuropathol. 2025. PMID: 40522345 Free PMC article.
-
Cerebral cortex swelling in V180I genetic Creutzfeldt-Jakob disease: comparative imaging study between sporadic and V180I genetic Creutzfeldt-Jakob disease in the early stage.Prion. 2023 Dec;17(1):105-110. doi: 10.1080/19336896.2023.2197809. Prion. 2023. PMID: 37013454 Free PMC article.
-
Human PrP E219K: a new and promising substrate for robust RT-QuIC amplification of human prions with potential for strain discrimination.Microbiol Spectr. 2025 Aug 5;13(8):e0029225. doi: 10.1128/spectrum.00292-25. Epub 2025 Jul 10. Microbiol Spectr. 2025. PMID: 40637424 Free PMC article.
-
Prions and prion diseases: Insights from the eye.Exp Eye Res. 2020 Oct;199:108200. doi: 10.1016/j.exer.2020.108200. Epub 2020 Aug 25. Exp Eye Res. 2020. PMID: 32858007 Free PMC article. Review.
-
A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis siné myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies.J Am Acad Dermatol. 2006 Apr;54(4):597-613. doi: 10.1016/j.jaad.2005.10.041. Epub 2006 Jan 23. J Am Acad Dermatol. 2006. PMID: 16546580
Cited by
-
What would it take to prove that a chronic infection is a causal agent in Alzheimer's disease?Trends Neurosci. 2025 Aug;48(8):608-623. doi: 10.1016/j.tins.2025.05.009. Epub 2025 Jun 16. Trends Neurosci. 2025. PMID: 40527696 Review.
-
Altered properties of amyloidogenic prion protein in genetic Creutzfeldt-Jakob disease with PRNP V180I mutation in response to pentosan polysulfate.Brain Pathol. 2023 Sep;33(5):e13197. doi: 10.1111/bpa.13197. Epub 2023 Jul 31. Brain Pathol. 2023. PMID: 37525413 Free PMC article.
References
-
- Ladogana A., Puopolo M., Croes E.A., Budka H., Jarius C., Collins S., Klug G.M., Sutcliffe T., Giulivi A., Alperovitch A., et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64:1586–1591. doi: 10.1212/01.WNL.0000160117.56690.B2. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- 20FC1054/Grant-in-Aid from the Research Committee of Prion Disease and Slow Virus Infection of the Ministry of Health, Labour, and Welfare of Japan
- 20FC2001/Grant-in-Aid from the Research Committee of Surveillance and Infection Control of Prion Disease of the Ministry of Health, Labour, and Welfare of Japan
LinkOut - more resources
Full Text Sources
Medical
Research Materials
