

Systematic Review of Clinical and Pathophysiological Features of Genetic Creutzfeldt-Jakob Disease Caused by a Val-to-Ile Mutation at Codon 180 in the Prion Protein Gene
- PMID: 36499498
- PMCID: PMC9737045
- DOI: 10.3390/ijms232315172
Systematic Review of Clinical and Pathophysiological Features of Genetic Creutzfeldt-Jakob Disease Caused by a Val-to-Ile Mutation at Codon 180 in the Prion Protein Gene
Abstract
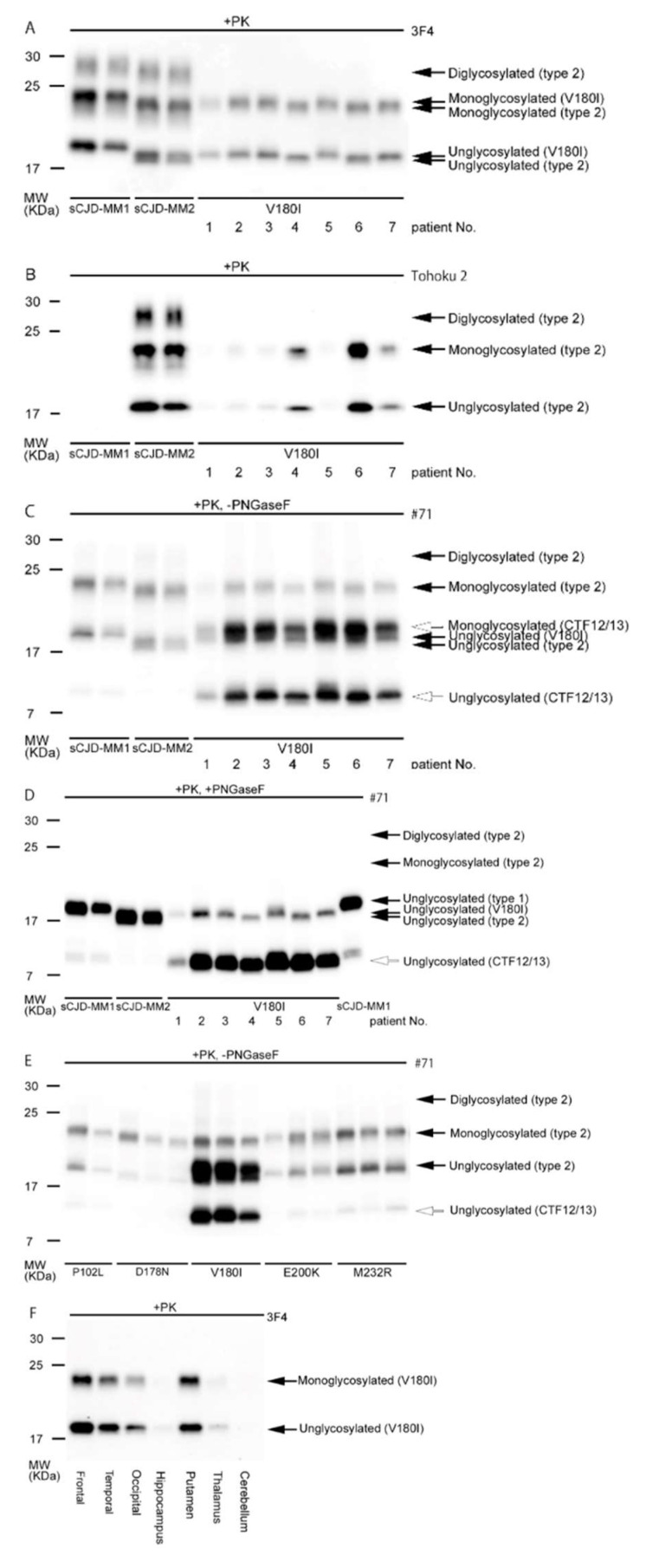
Genetic Creutzfeldt-Jakob disease (gCJD) is a subtype of genetic prion diseases (gPrDs) caused by the accumulation of mutated pathological prion proteins (PrPSc). gCJD has a phenotypic similarity with sporadic CJD (sCJD). In Japan, gCJD with a Val to Ile substitution at codon 180 (V180I-gCJD) is the most frequent gPrD, while the mutation is extremely rare in countries other than Japan and Korea. In this article, we aim to review previously elucidated clinical and biochemical features of V180I-gCJD, expecting to advance the understanding of this unique subtype in gCJD. Compared to classical sCJD, specific clinical features of V180I-gCJD include older age at onset, a relatively slow progression of dementia, and a lower positivity for developing myoclonus, cerebellar, pyramidal signs, and visual disturbance. Diffuse edematous ribboning hyperintensity of the cerebral cortex, without occipital lobes in diffusion-weighted magnetic resonance imaging, is also specific. Laboratory data reveal the low positivity of PrPSc in the cerebrospinal fluid and periodic sharp wave complexes on an electroencephalogram. Most patients with V180I-gCJD have been reported to have no family history, probably due to the older age at onset, and clinical and biochemical features indicate the specific phenotype associated with the prion protein gene mutation.
Keywords: V180I; Val-to-Ile substitution at codon 180; genetic Creutzfeldt–Jakob disease; genetic prion disease; normal prion proteins; pathological prion proteins; prion protein gene.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Ladogana A., Puopolo M., Croes E.A., Budka H., Jarius C., Collins S., Klug G.M., Sutcliffe T., Giulivi A., Alperovitch A., et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64:1586–1591. doi: 10.1212/01.WNL.0000160117.56690.B2. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- 20FC1054/Grant-in-Aid from the Research Committee of Prion Disease and Slow Virus Infection of the Ministry of Health, Labour, and Welfare of Japan
- 20FC2001/Grant-in-Aid from the Research Committee of Surveillance and Infection Control of Prion Disease of the Ministry of Health, Labour, and Welfare of Japan
LinkOut - more resources
Full Text Sources
Medical
Research Materials
