

Tetrel Bonding in Anion Recognition: A First Principles Investigation
- PMID: 36500544
- PMCID: PMC9738195
- DOI: 10.3390/molecules27238449
Tetrel Bonding in Anion Recognition: A First Principles Investigation
Abstract
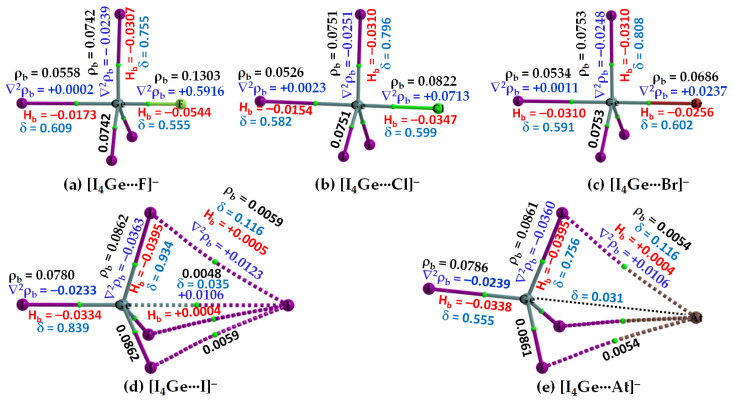
Twenty-five molecule-anion complex systems [I4Tt···X-] (Tt = C, Si, Ge, Sn and Pb; X = F, Cl, Br, I and At) were examined using density functional theory (ωB97X-D) and ab initio (MP2 and CCSD) methods to demonstrate the ability of the tetrel atoms in molecular entities, I4Tt, to recognize the halide anions when in close proximity. The tetrel bond strength for the [I4C···X-] series and [I4Tt···X-] (Tt = Si, Sn; X = I, At), was weak-to-moderate, whereas that in the remaining 16 complexes was dative tetrel bond type with very large interaction energies and short Tt···X close contact distances. The basis set superposition error corrected interaction energies calculated with the highest-level theory applied, [CCSD(T)/def2-TZVPPD], ranged from -3.0 to -112.2 kcal mol-1. The significant variation in interaction energies was realized as a result of different levels of tetrel bonding environment between the interacting partners at the equilibrium geometries of the complex systems. Although the ωB97X-D computed intermolecular geometries and interaction energies of a majority of the [I4Tt···X-] complexes were close to those predicted by the highest level of theory, the MP2 results were shown to be misleading for some of these systems. To provide insight into the nature of the intermolecular chemical bonding environment in the 25 molecule-anion complexes investigated, we discussed the charge-density-based topological and isosurface features that emanated from the application of the quantum theory of atoms in molecules and independent gradient model approaches, respectively.
Keywords: MESP analysis; anion recognition; charge-density analysis; chemical bonding; dative bond formation; first-principles calculations; non-covalent interactions; tetrel bond; weak-to-strong interaction energy.
Conflict of interest statement
The author declares no conflict of interest. The funders had absolutely no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures











Similar articles
-
Methylammonium Tetrel Halide Perovskite Ion Pairs and Their Dimers: The Interplay between the Hydrogen-, Pnictogen- and Tetrel-Bonding Interactions.Int J Mol Sci. 2023 Jun 23;24(13):10554. doi: 10.3390/ijms241310554. Int J Mol Sci. 2023. PMID: 37445738 Free PMC article.
-
The Tetrel Bond and Tetrel Halide Perovskite Semiconductors.Int J Mol Sci. 2023 Apr 3;24(7):6659. doi: 10.3390/ijms24076659. Int J Mol Sci. 2023. PMID: 37047632 Free PMC article.
-
Strong Tetrel Bonds: Theoretical Aspects and Experimental Evidence.Molecules. 2018 Oct 15;23(10):2642. doi: 10.3390/molecules23102642. Molecules. 2018. PMID: 30326582 Free PMC article.
-
Calculations on noncovalent interactions and databases of benchmark interaction energies.Acc Chem Res. 2012 Apr 17;45(4):663-72. doi: 10.1021/ar200255p. Epub 2012 Jan 6. Acc Chem Res. 2012. PMID: 22225511 Review.
-
Ab initio study of hydrogen-bond formation between aliphatic and phenolic hydroxy groups and selected amino acid side chains.J Phys Chem A. 2008 May 8;112(18):4342-54. doi: 10.1021/jp7108847. Epub 2008 Mar 29. J Phys Chem A. 2008. PMID: 18373368
Cited by
-
Halogen Bond via an Electrophilic π-Hole on Halogen in Molecules: Does It Exist?Int J Mol Sci. 2024 Apr 23;25(9):4587. doi: 10.3390/ijms25094587. Int J Mol Sci. 2024. PMID: 38731806 Free PMC article.
-
Search for Osme Bonds with π Systems as Electron Donors.Molecules. 2023 Dec 22;29(1):79. doi: 10.3390/molecules29010079. Molecules. 2023. PMID: 38202661 Free PMC article.
-
Synthesis, Structural Characterization, and Theoretical Analysis of Nonconventional Bonding in Dinuclear Zinc(II) Complexes with Tridentate Schiff Bases.ACS Omega. 2024 Sep 27;9(40):41787-41796. doi: 10.1021/acsomega.4c06136. eCollection 2024 Oct 8. ACS Omega. 2024. PMID: 39398134 Free PMC article.
-
Methylammonium Tetrel Halide Perovskite Ion Pairs and Their Dimers: The Interplay between the Hydrogen-, Pnictogen- and Tetrel-Bonding Interactions.Int J Mol Sci. 2023 Jun 23;24(13):10554. doi: 10.3390/ijms241310554. Int J Mol Sci. 2023. PMID: 37445738 Free PMC article.
-
The Tetrel Bond and Tetrel Halide Perovskite Semiconductors.Int J Mol Sci. 2023 Apr 3;24(7):6659. doi: 10.3390/ijms24076659. Int J Mol Sci. 2023. PMID: 37047632 Free PMC article.
References
-
- Lim J.Y.C., Beer P.D. Sigma-Hole Interactions in Anion Recognition. Chem. 2018;4:731–783. doi: 10.1016/j.chempr.2018.02.022. - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
