

Novel machine learning approaches revolutionize protein knowledge
- PMID: 36504138
- PMCID: PMC10570143
- DOI: 10.1016/j.tibs.2022.11.001
Novel machine learning approaches revolutionize protein knowledge
Abstract
Breakthrough methods in machine learning (ML), protein structure prediction, and novel ultrafast structural aligners are revolutionizing structural biology. Obtaining accurate models of proteins and annotating their functions on a large scale is no longer limited by time and resources. The most recent method to be top ranked by the Critical Assessment of Structure Prediction (CASP) assessment, AlphaFold 2 (AF2), is capable of building structural models with an accuracy comparable to that of experimental structures. Annotations of 3D models are keeping pace with the deposition of the structures due to advancements in protein language models (pLMs) and structural aligners that help validate these transferred annotations. In this review we describe how recent developments in ML for protein science are making large-scale structural bioinformatics available to the general scientific community.
Keywords: AI; AlphaFold2; embeddings; machine learning; pLM; protein structure prediction; structure alignment.
Copyright © 2022 The Author(s). Published by Elsevier Ltd.. All rights reserved.
Conflict of interest statement
Declaration of interests No interests are declared.
Figures

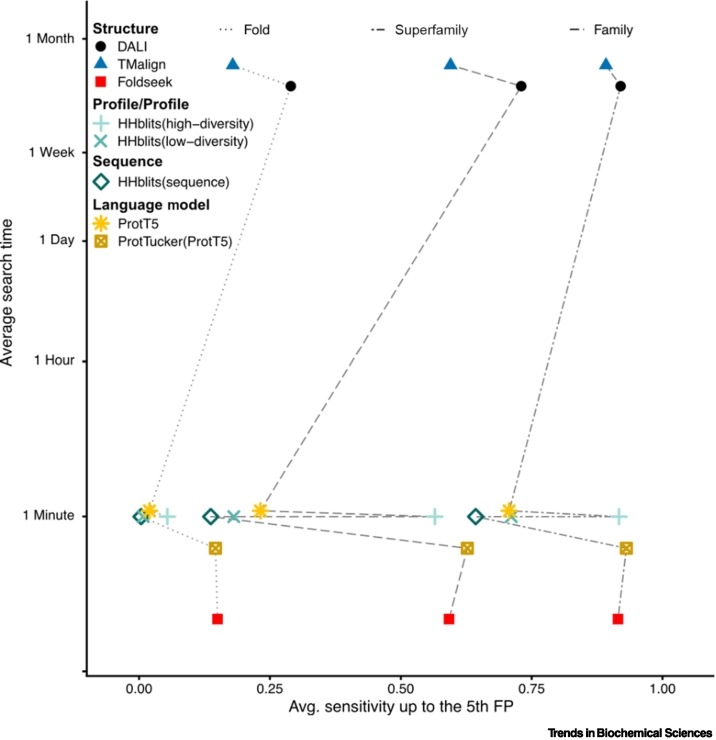
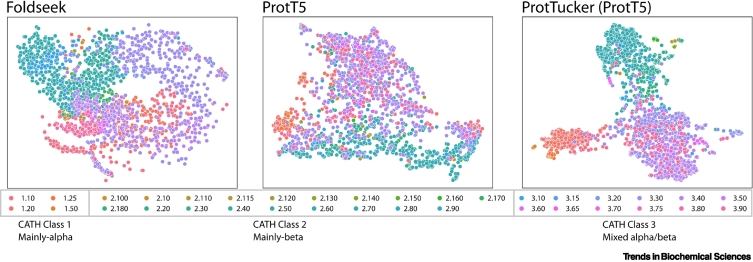
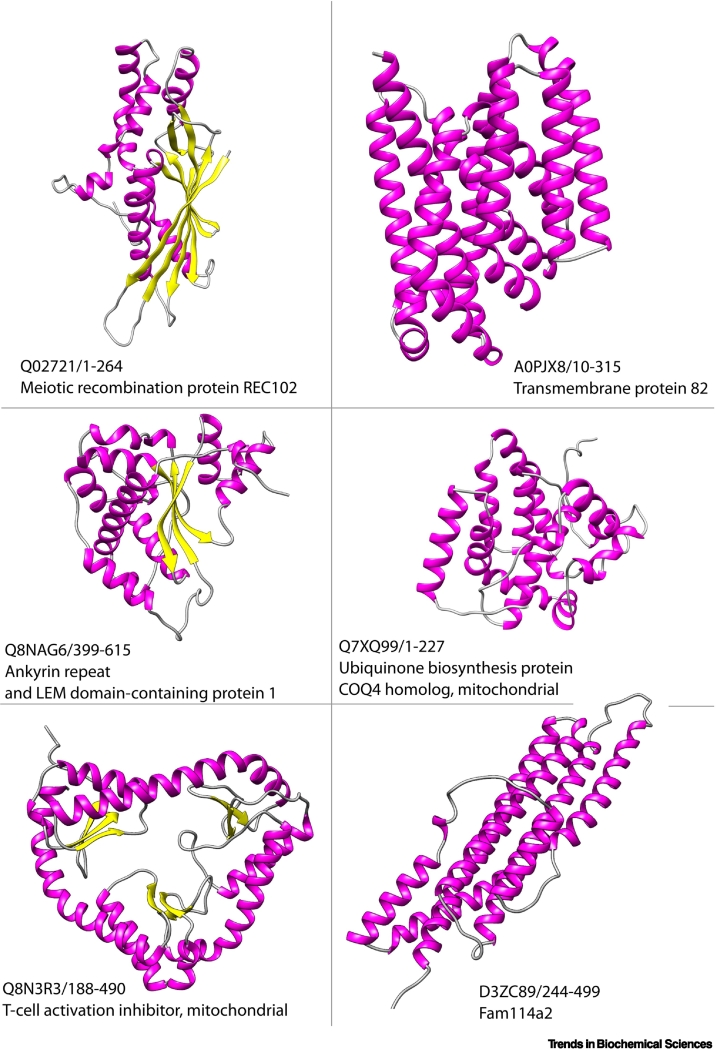
References
-
- Liu J., Rost B. CHOP proteins into structural domain-like fragments. Proteins. 2004;55:678–688. - PubMed
-
- Orengo C.A., Thornton J.M. Protein families and their evolution—a structural perspective. Annu. Rev. Biochem. 2005;74:867–900. - PubMed
-
- Chothia C. Proteins. One thousand families for the molecular biologist. Nature. 1992;357:543–544. - PubMed
-
- Orengo C.A., et al. Protein superfamilies and domain superfolds. Nature. 1994;372:631–634. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
