

Activation of HIV-1 proviruses increases downstream chromatin accessibility
- PMID: 36505924
- PMCID: PMC9732416
- DOI: 10.1016/j.isci.2022.105490
Activation of HIV-1 proviruses increases downstream chromatin accessibility
Abstract
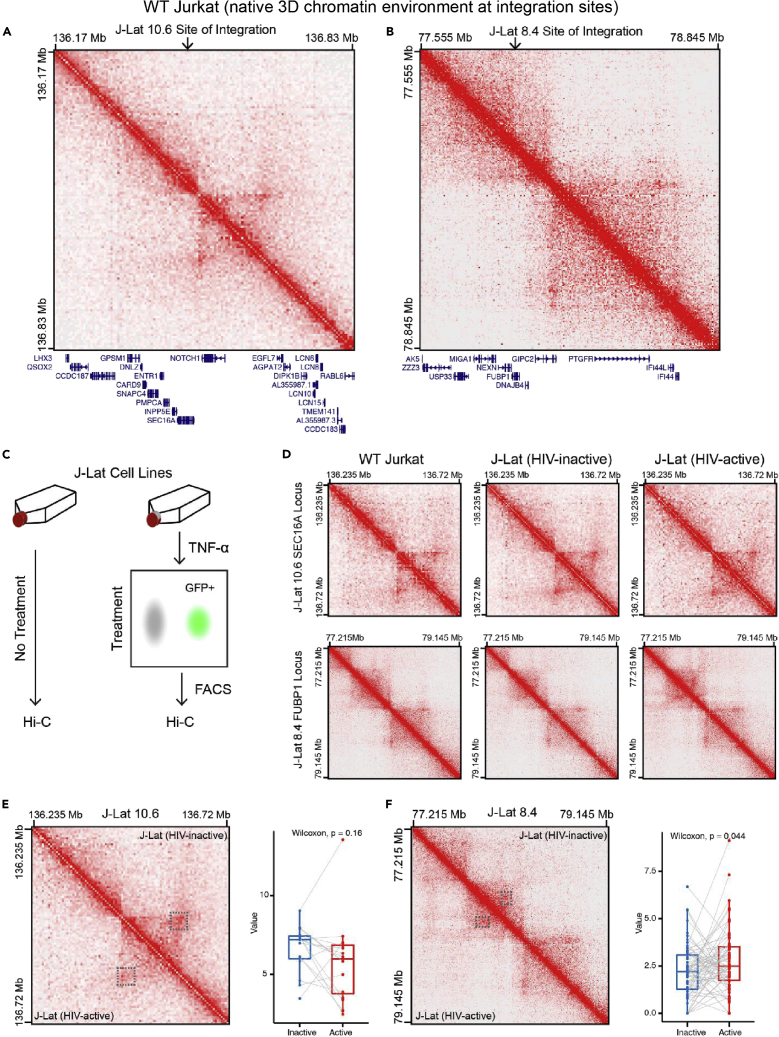
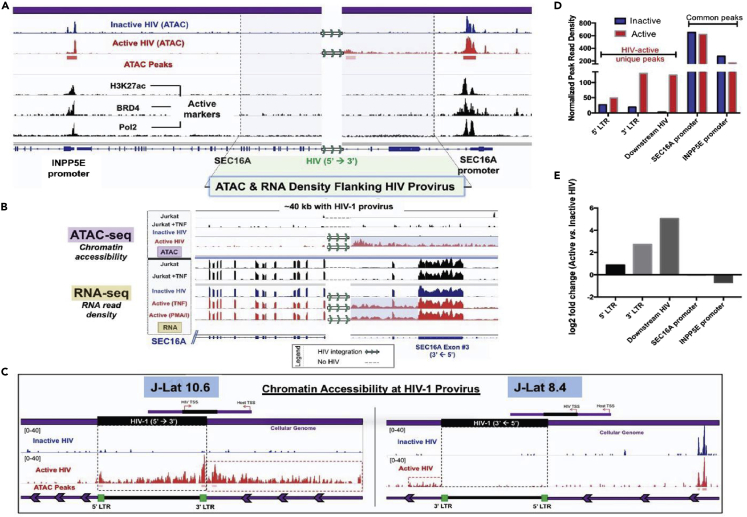
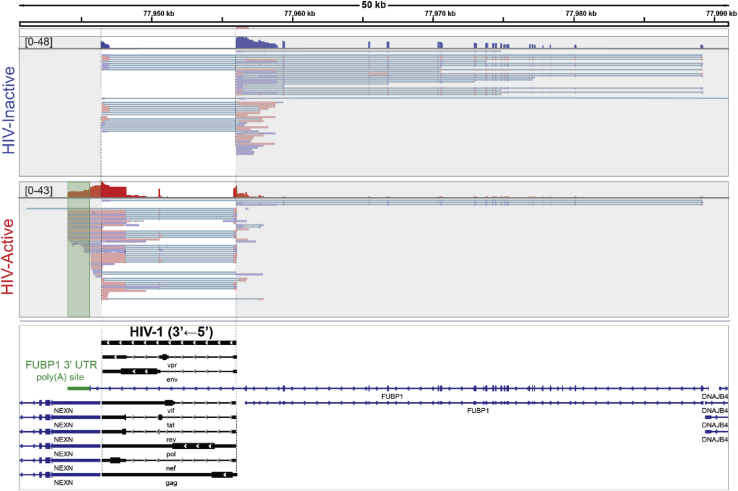
It is unclear how the activation of HIV-1 transcription affects chromatin structure. We interrogated chromatin organization both genome-wide and nearby HIV-1 integration sites using Hi-C and ATAC-seq. In conjunction, we analyzed the transcription of the HIV-1 genome and neighboring genes. We found that long-range chromatin contacts did not differ significantly between uninfected cells and those harboring an integrated HIV-1 genome, whether the HIV-1 genome was actively transcribed or inactive. Instead, the activation of HIV-1 transcription changes chromatin accessibility immediately downstream of the provirus, demonstrating that HIV-1 can alter local cellular chromatin structure. Finally, we examined HIV-1 and neighboring host gene transcripts with long-read sequencing and found populations of chimeric RNAs both virus-to-host and host-to-virus. Thus, multiomics profiling revealed that the activation of HIV-1 transcription led to local changes in chromatin organization and altered the expression of neighboring host genes.
Keywords: Biological sciences; Chromosome organization; Molecular biology; Molecular interaction.
© 2022.
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
Integration site-dependent HIV-1 promoter activity shapes host chromatin conformation.Genome Res. 2023 Jun;33(6):891-906. doi: 10.1101/gr.277698.123. Epub 2023 Jun 9. Genome Res. 2023. PMID: 37295842 Free PMC article.
-
Proviruses with Long-Term Stable Expression Accumulate in Transcriptionally Active Chromatin Close to the Gene Regulatory Elements: Comparison of ASLV-, HIV- and MLV-Derived Vectors.Viruses. 2018 Mar 8;10(3):116. doi: 10.3390/v10030116. Viruses. 2018. PMID: 29517993 Free PMC article.
-
Computational Integration of HSV-1 Multi-omics Data.Methods Mol Biol. 2023;2610:31-48. doi: 10.1007/978-1-0716-2895-9_3. Methods Mol Biol. 2023. PMID: 36534279 Review.
-
Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy.J Clin Invest. 2019 Mar 1;129(3):988-998. doi: 10.1172/JCI124291. Epub 2019 Jan 28. J Clin Invest. 2019. PMID: 30688658 Free PMC article. Clinical Trial.
-
The regulation of HIV-1 gene expression: the emerging role of chromatin.DNA Cell Biol. 2002 Oct;21(10):697-705. doi: 10.1089/104454902760599672. DNA Cell Biol. 2002. PMID: 12443539 Review.
Cited by
-
Impact of chromatin on HIV-1 latency: a multi-dimensional perspective.Epigenetics Chromatin. 2025 Mar 8;18(1):9. doi: 10.1186/s13072-025-00573-x. Epigenetics Chromatin. 2025. PMID: 40055755 Free PMC article. Review.
-
Disruption of LEDGF/p75-directed integration derepresses antisense transcription of the HIV-1 genome.bioRxiv [Preprint]. 2024 Dec 6:2024.12.06.627169. doi: 10.1101/2024.12.06.627169. bioRxiv. 2024. PMID: 39677798 Free PMC article. Preprint.
-
Integrated ATAC-seq and RNA-seq data analysis identifies transcription factors related to rice stripe virus infection in Oryza sativa.Mol Plant Pathol. 2024 Mar;25(3):e13446. doi: 10.1111/mpp.13446. Mol Plant Pathol. 2024. PMID: 38502176 Free PMC article.
-
Viral remodeling of the 4D nucleome.Exp Mol Med. 2024 Apr;56(4):799-808. doi: 10.1038/s12276-024-01207-0. Epub 2024 Apr 25. Exp Mol Med. 2024. PMID: 38658699 Free PMC article. Review.
-
The p400 complex promotes HIV-1 latency by suppressing viral transcription and altering the host cell state.Nucleic Acids Res. 2025 Aug 11;53(15):gkaf764. doi: 10.1093/nar/gkaf764. Nucleic Acids Res. 2025. PMID: 40842241 Free PMC article.
References
-
- Chun T.W., Engel D., Mizell S.B., Hallahan C.W., Fischette M., Park S., Davey R.T., Jr., Dybul M., Kovacs J.A., Metcalf J.A., et al. Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat. Med. 1999;5:651–655. - PubMed
-
- Sonza S., Mutimer H.P., Oelrichs R., Jardine D., Harvey K., Dunne A., Purcell D.F., Birch C., Crowe S.M. Monocytes harbour replication-competent, non-latent HIV-1 in patients on highly active antiretroviral therapy. AIDS. 2001;15:17–22. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
