

Herpes Simplex Virus 1-Induced Ferroptosis Contributes to Viral Encephalitis
- PMID: 36507835
- PMCID: PMC9973258
- DOI: 10.1128/mbio.02370-22
Herpes Simplex Virus 1-Induced Ferroptosis Contributes to Viral Encephalitis
Abstract
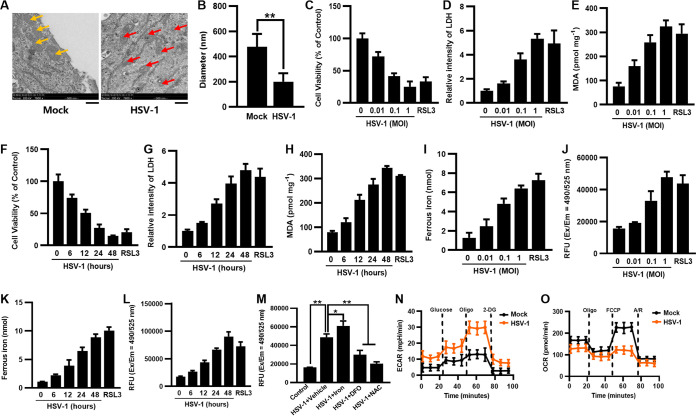
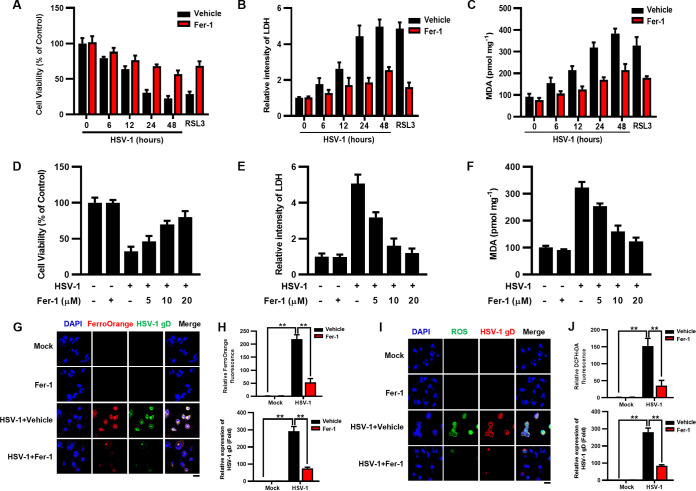
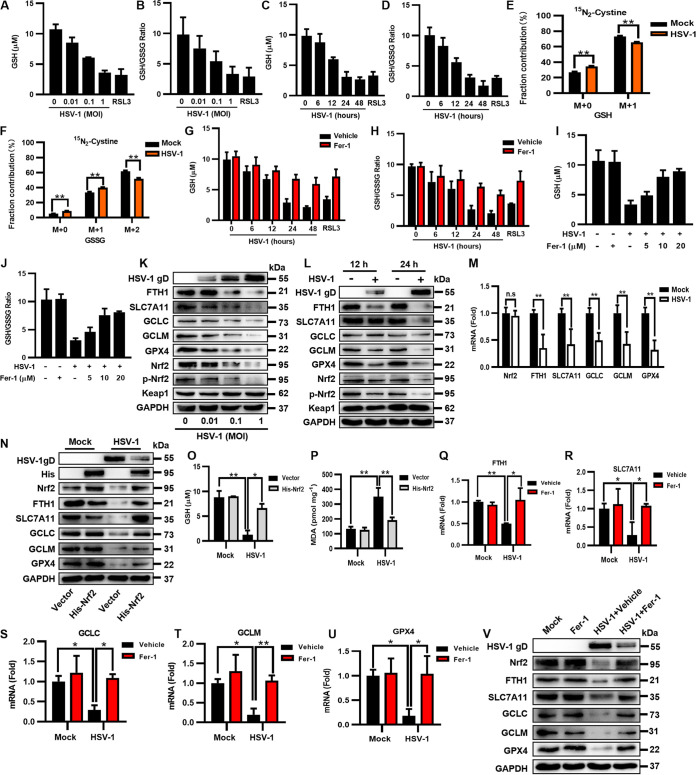
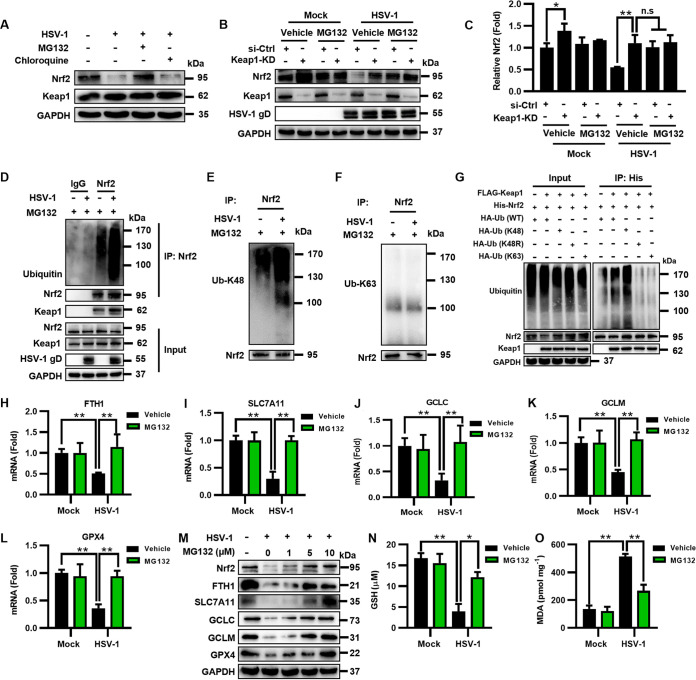
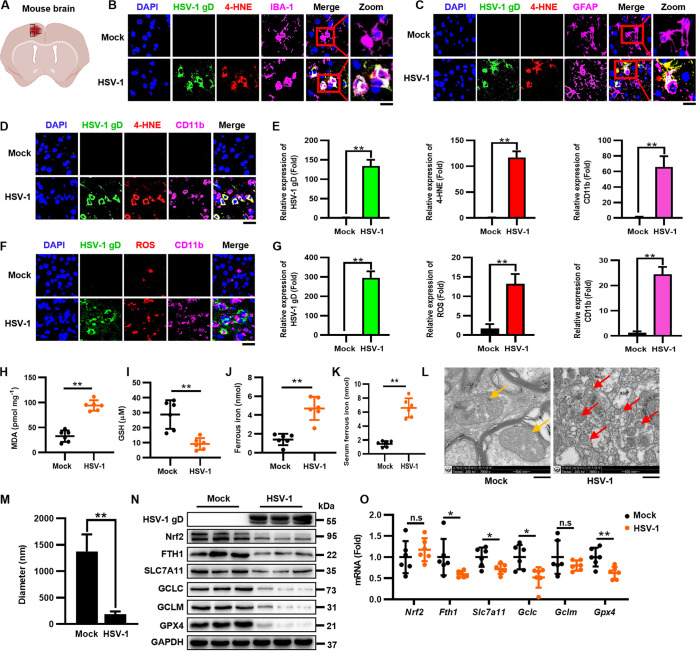
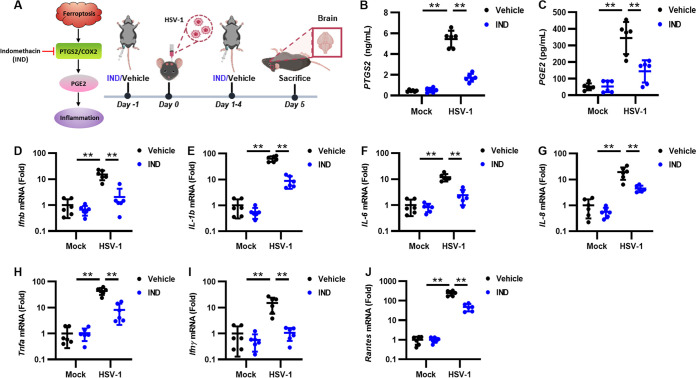
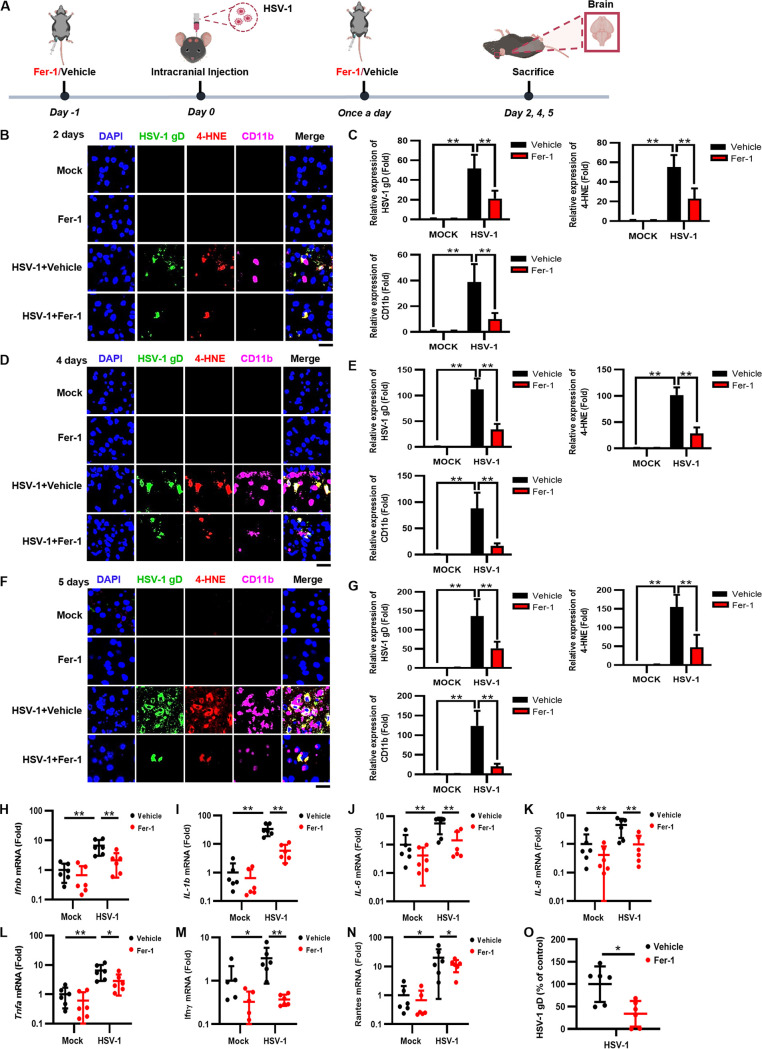
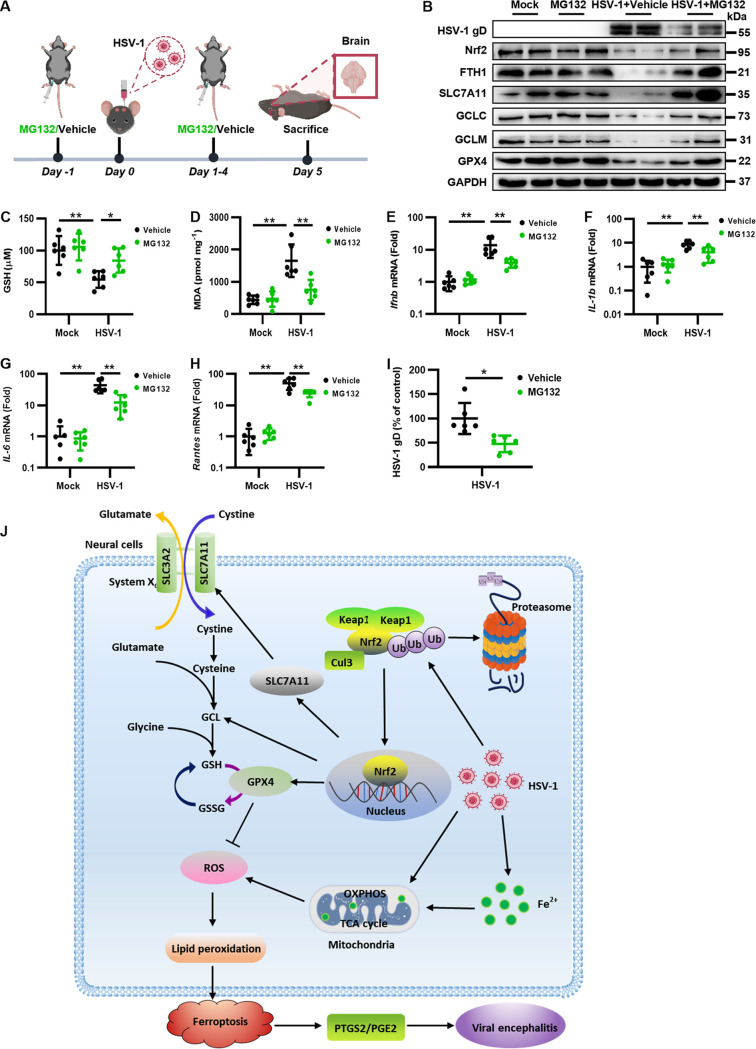
Herpes simplex virus 1 (HSV-1) is a DNA virus belonging to the family Herpesviridae. HSV-1 infection causes severe neurological disease in the central nervous system (CNS), including encephalitis. Ferroptosis is a nonapoptotic form of programmed cell death that contributes to different neurological inflammatory diseases. However, whether HSV-1 induces ferroptosis in the CNS and the role of ferroptosis in viral pathogenesis remain unclear. Here, we demonstrate that HSV-1 induces ferroptosis, as hallmarks of ferroptosis, including Fe2+ overload, reactive oxygen species (ROS) accumulation, glutathione (GSH) depletion, lipid peroxidation, and mitochondrion shrinkage, are observed in HSV-1-infected cultured human astrocytes, microglia cells, and murine brains. Moreover, HSV-1 infection enhances the E3 ubiquitin ligase Keap1 (Kelch-like ECH-related protein 1)-mediated ubiquitination and degradation of nuclear factor E2-related factor 2 (Nrf2), a transcription factor that regulates the expression of antioxidative genes, thereby disturbing cellular redox homeostasis and promoting ferroptosis. Furthermore, HSV-1-induced ferroptosis is tightly associated with the process of viral encephalitis in a mouse model, and the ferroptosis-activated upregulation of prostaglandin-endoperoxide synthase 2 (PTGS2) and prostaglandin E2 (PGE2) plays an important role in HSV-1-caused inflammation and encephalitis. Importantly, the inhibition of ferroptosis by a ferroptosis inhibitor or a proteasome inhibitor to suppress Nrf2 degradation effectively alleviated HSV-1 encephalitis. Together, our findings demonstrate the interaction between HSV-1 infection and ferroptosis and provide novel insights into the pathogenesis of HSV-1 encephalitis. IMPORTANCE Ferroptosis is a nonapoptotic form of programmed cell death that contributes to different neurological inflammatory diseases. However, whether HSV-1 induces ferroptosis in the CNS and the role of ferroptosis in viral pathogenesis remain unclear. In the current study, we demonstrate that HSV-1 infection induces ferroptosis, as Fe2+ overload, ROS accumulation, GSH depletion, lipid peroxidation, and mitochondrion shrinkage, all of which are hallmarks of ferroptosis, are observed in human cultured astrocytes, microglia cells, and murine brains infected with HSV-1. Moreover, HSV-1 infection enhances Keap1-dependent Nrf2 ubiquitination and degradation, which results in substantial reductions in the expression levels of antiferroptotic genes downstream of Nrf2, thereby disturbing cellular redox homeostasis and promoting ferroptosis. Furthermore, HSV-1-induced ferroptosis is tightly associated with the process of viral encephalitis in a mouse model, and the ferroptosis-activated upregulation of PTGS2 and PGE2 plays an important role in HSV-1-caused inflammation and encephalitis. Importantly, the inhibition of ferroptosis by either a ferroptosis inhibitor or a proteasome inhibitor to suppress HSV-1-induced Nrf2 degradation effectively alleviates HSV-1-caused neuro-damage and inflammation in infected mice. Overall, our findings uncover the interaction between HSV-1 infection and ferroptosis, shed novel light on the physiological impacts of ferroptosis on the pathogenesis of HSV-1 infection and encephalitis, and provide a promising therapeutic strategy to treat this important infectious disease with a worldwide distribution.
Keywords: HSV-1; Nrf2-Keap1; PTGS2/PGE2; ferroptosis; viral encephalitis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- WHO. 2022. Herpes simplex virus. WHO, Geneva, Switzerland. https://www.who.int/news-room/fact-sheets/detail/herpes-simplex-virus.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
