

Synthetical lethality of Werner helicase and mismatch repair deficiency is mediated by p53 and PUMA in colon cancer
- PMID: 36508676
- PMCID: PMC9907101
- DOI: 10.1073/pnas.2211775119
Synthetical lethality of Werner helicase and mismatch repair deficiency is mediated by p53 and PUMA in colon cancer
Erratum in
-
Correction for Hao et al., Synthetical lethality of Werner helicase and mismatch repair deficiency is mediated by p53 and PUMA in colon cancer.Proc Natl Acad Sci U S A. 2025 Nov 25;122(47):e2529649122. doi: 10.1073/pnas.2529649122. Epub 2025 Nov 19. Proc Natl Acad Sci U S A. 2025. PMID: 41259151 Free PMC article. No abstract available.
Abstract
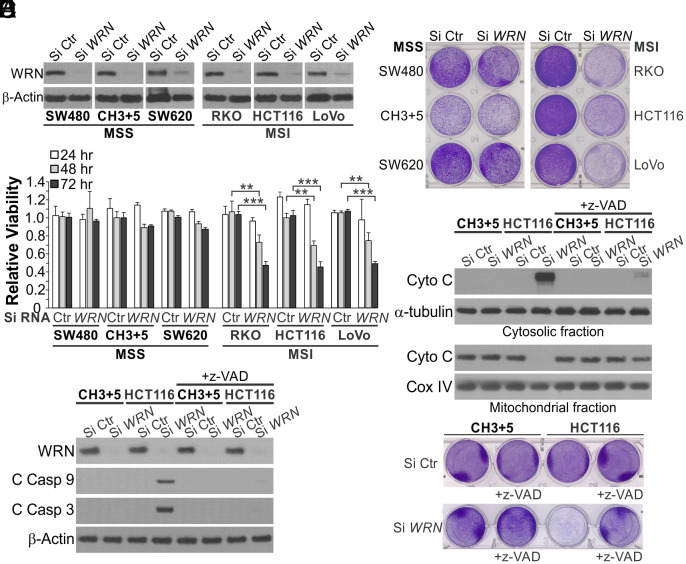
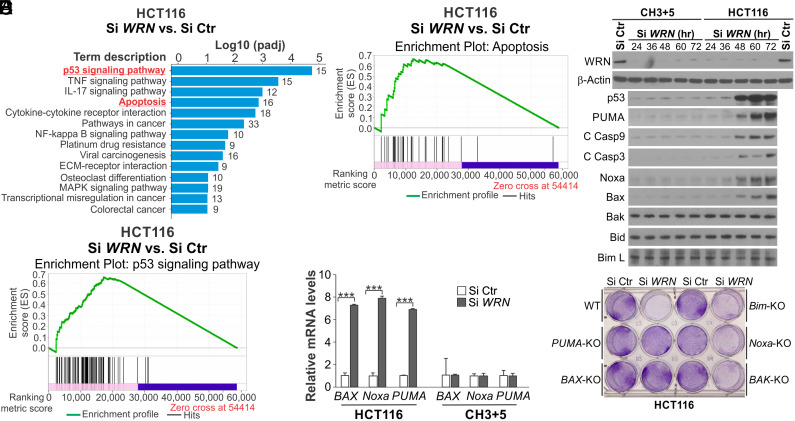
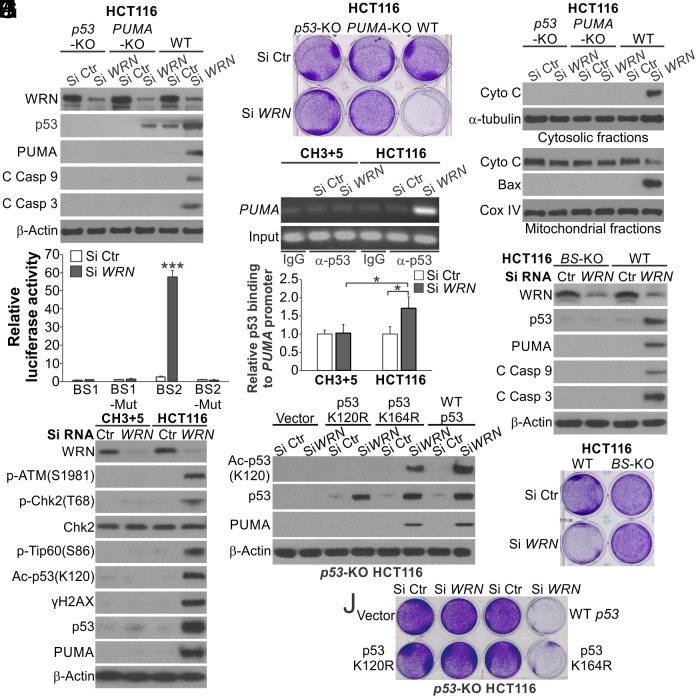
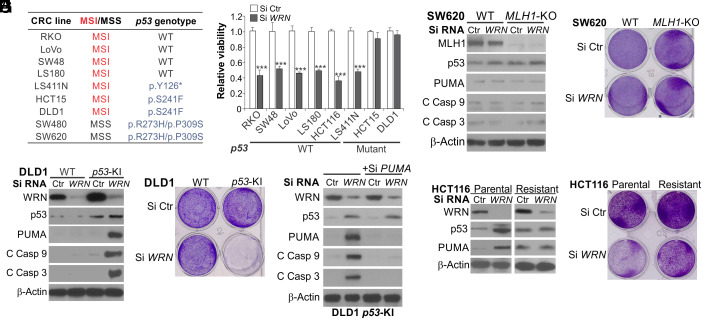
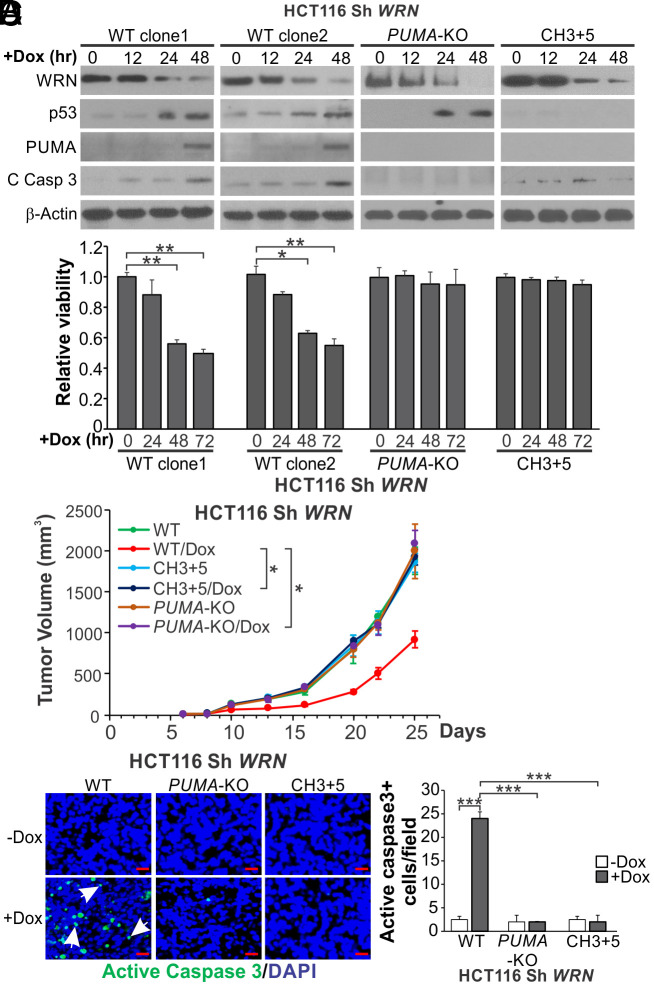
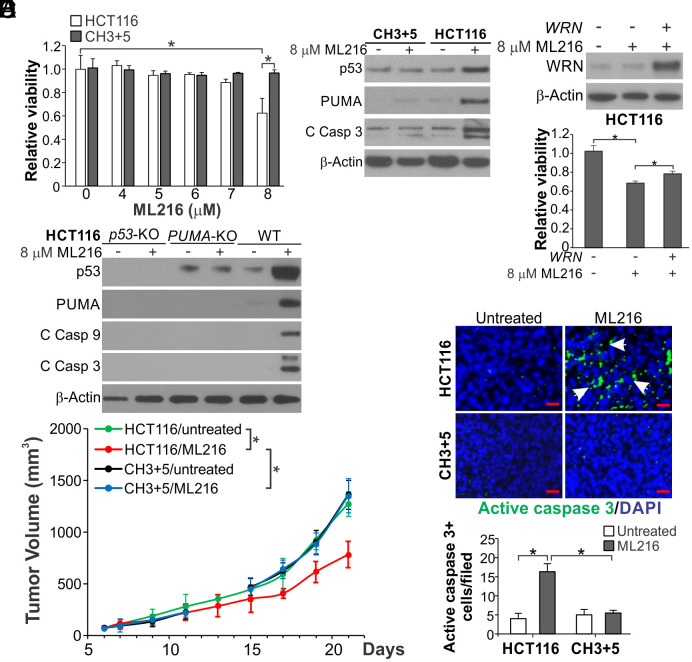
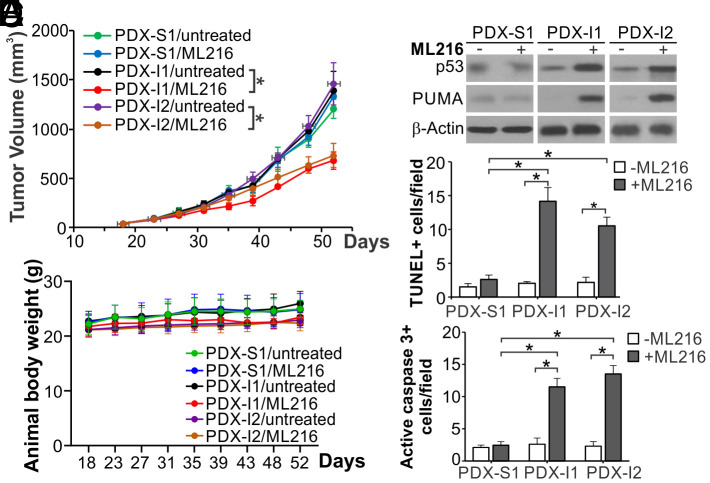
Synthetic lethality is a powerful approach for targeting oncogenic drivers in cancer. Recent studies revealed that cancer cells with microsatellite instability (MSI) require Werner (WRN) helicase for survival; however, the underlying mechanism remains unclear. In this study, we found that WRN depletion strongly induced p53 and its downstream apoptotic target PUMA in MSI colorectal cancer (CRC) cells. p53 or PUMA deletion abolished apoptosis induced by WRN depletion in MSI CRC cells. Importantly, correction of MSI abrogated the activation of p53/PUMA and cell killing, while induction of MSI led to sensitivity in isogenic CRC cells. Rare p53-mutant MSI CRC cells are resistant to WRN depletion due to lack of PUMA induction, which could be restored by wildtype (WT) p53 knock in or reconstitution. WRN depletion or treatment with the RecQ helicase inhibitor ML216 suppressed in vitro and in vivo growth of MSI CRCs in a p53/PUMA-dependent manner. ML216 treatment was efficacious in MSI CRC patient-derived xenografts. Interestingly, p53 gene remains WT in the majority of MSI CRCs. These results indicate a critical role of p53/PUMA-mediated apoptosis in the vulnerability of MSI CRCs to WRN loss, and support WRN as a promising therapeutic target in p53-WT MSI CRCs.
Keywords: PUMA; Werner; mismatch repair; p53; synthetic lethal.
Conflict of interest statement
The authors declare no competing interest.
Figures
Comment in
-
WRN suppresses p53/PUMA-induced apoptosis in colorectal cancer with microsatellite instability/mismatch repair deficiency.Proc Natl Acad Sci U S A. 2023 Jan 10;120(2):e2219963120. doi: 10.1073/pnas.2219963120. Epub 2023 Jan 4. Proc Natl Acad Sci U S A. 2023. PMID: 36598947 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
