

DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits
- PMID: 36510025
- PMCID: PMC10249665
- DOI: 10.1038/s41588-022-01248-z
DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits
Abstract
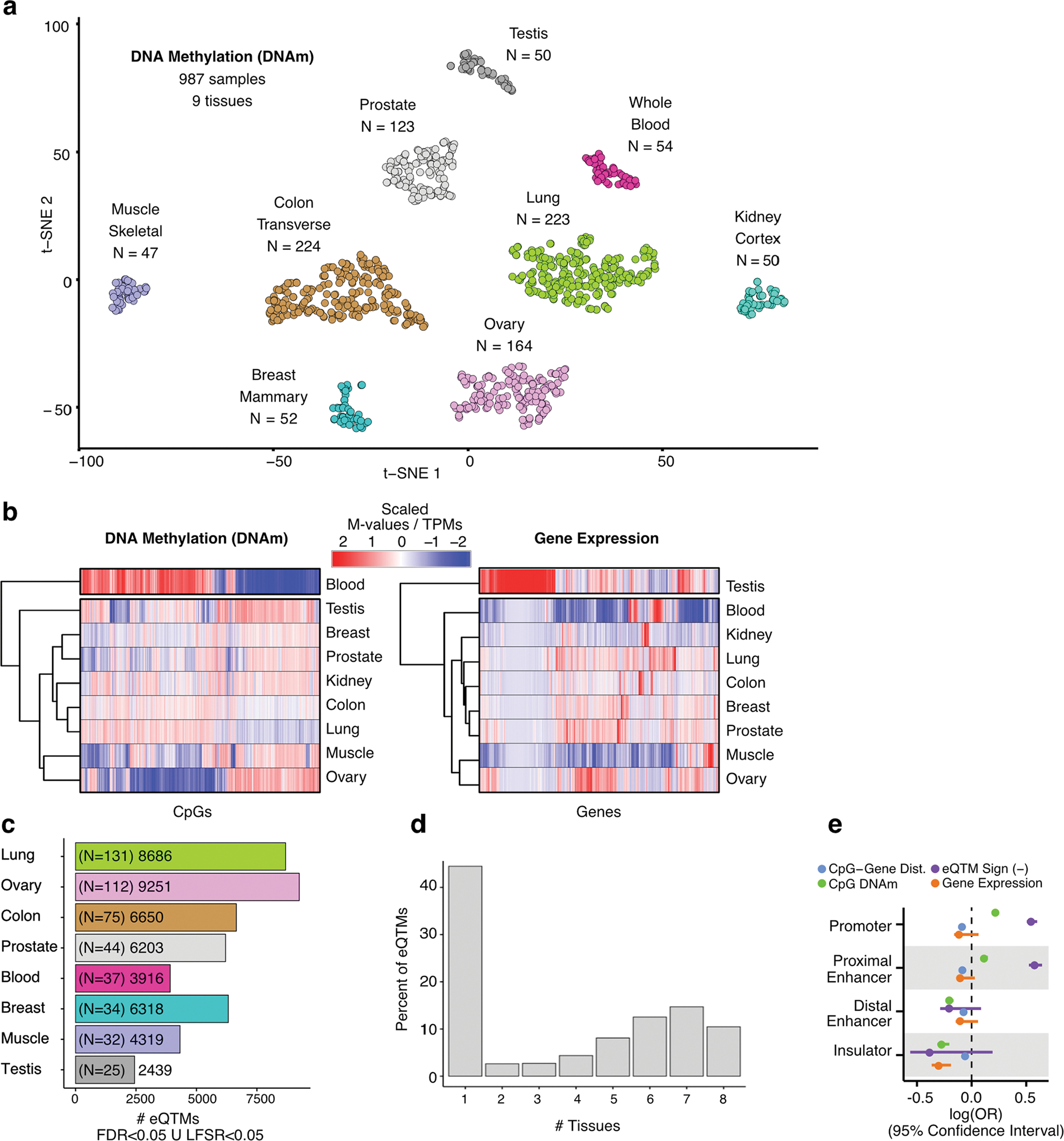
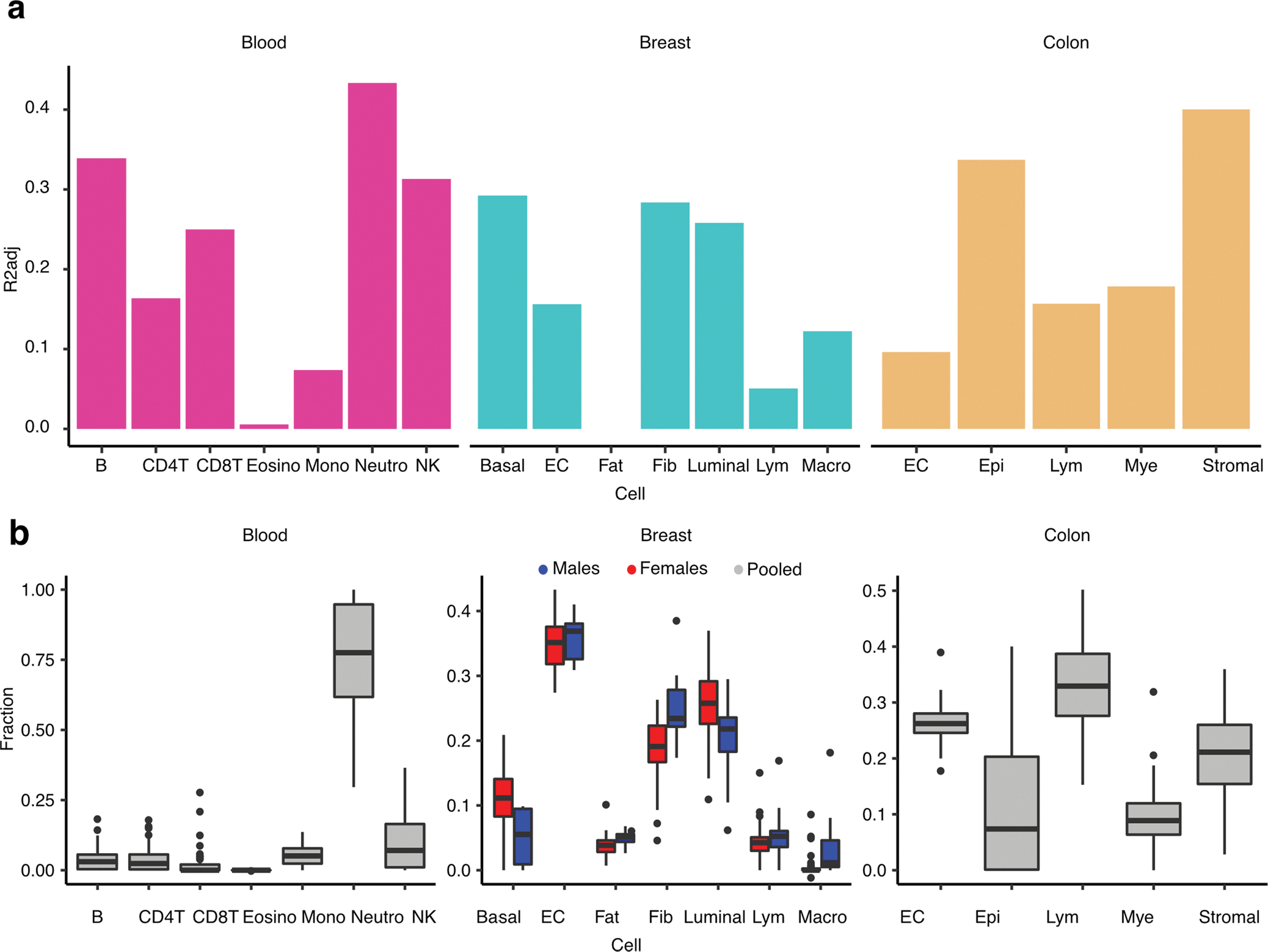
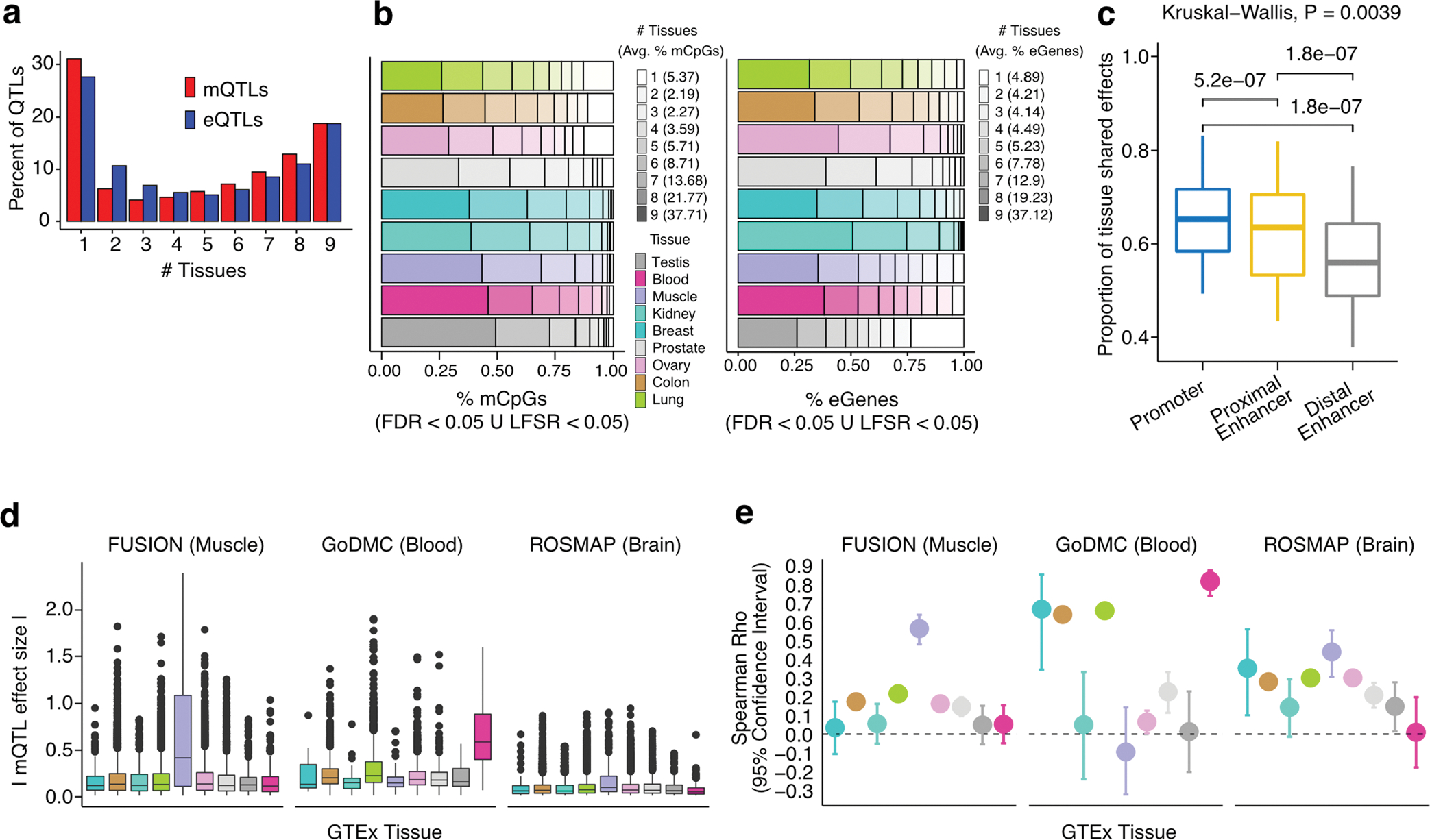
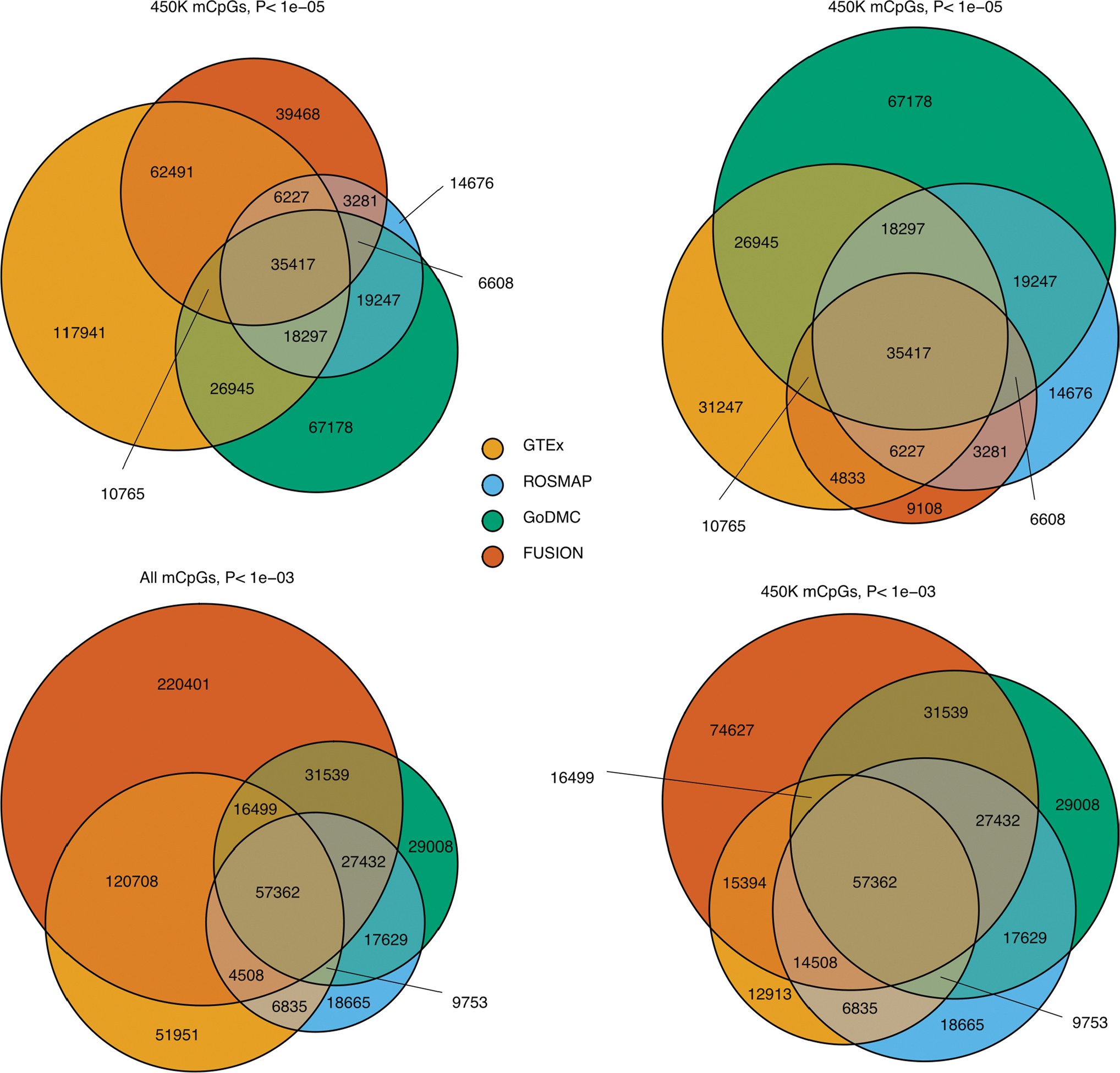
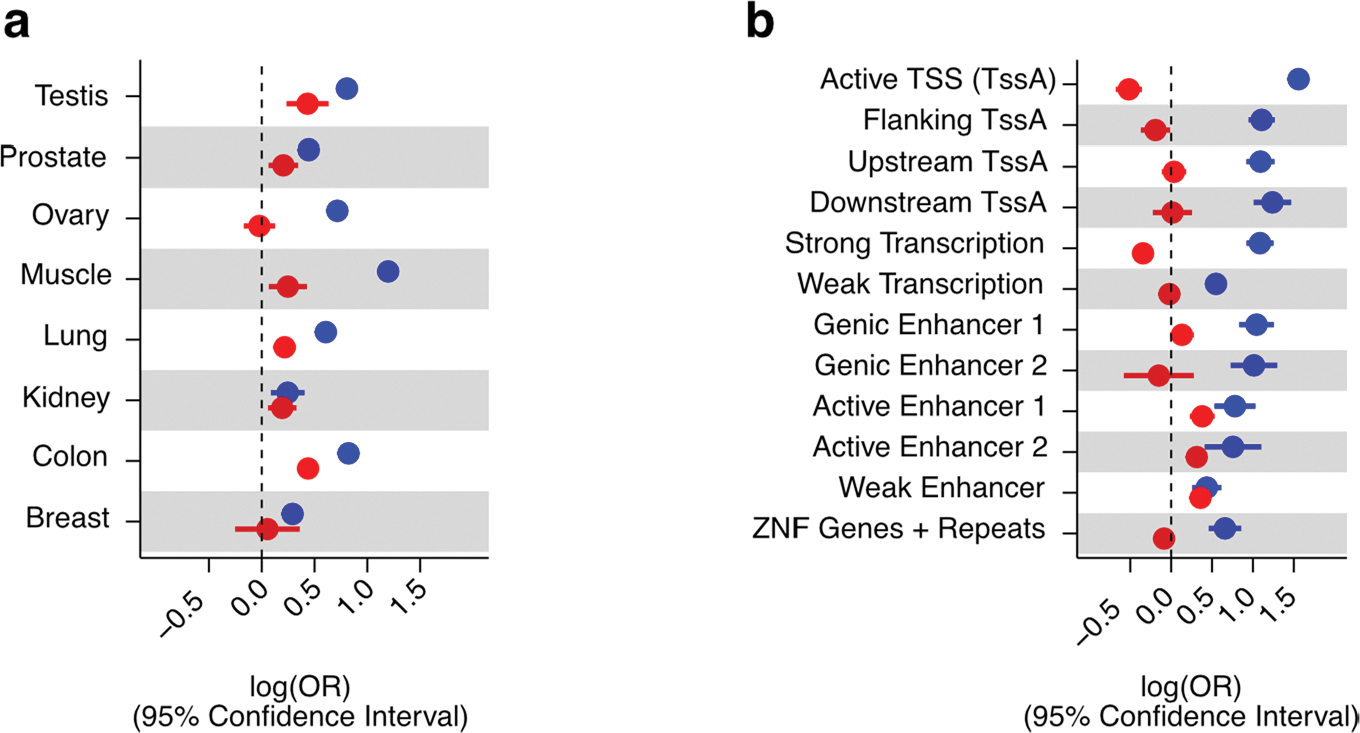
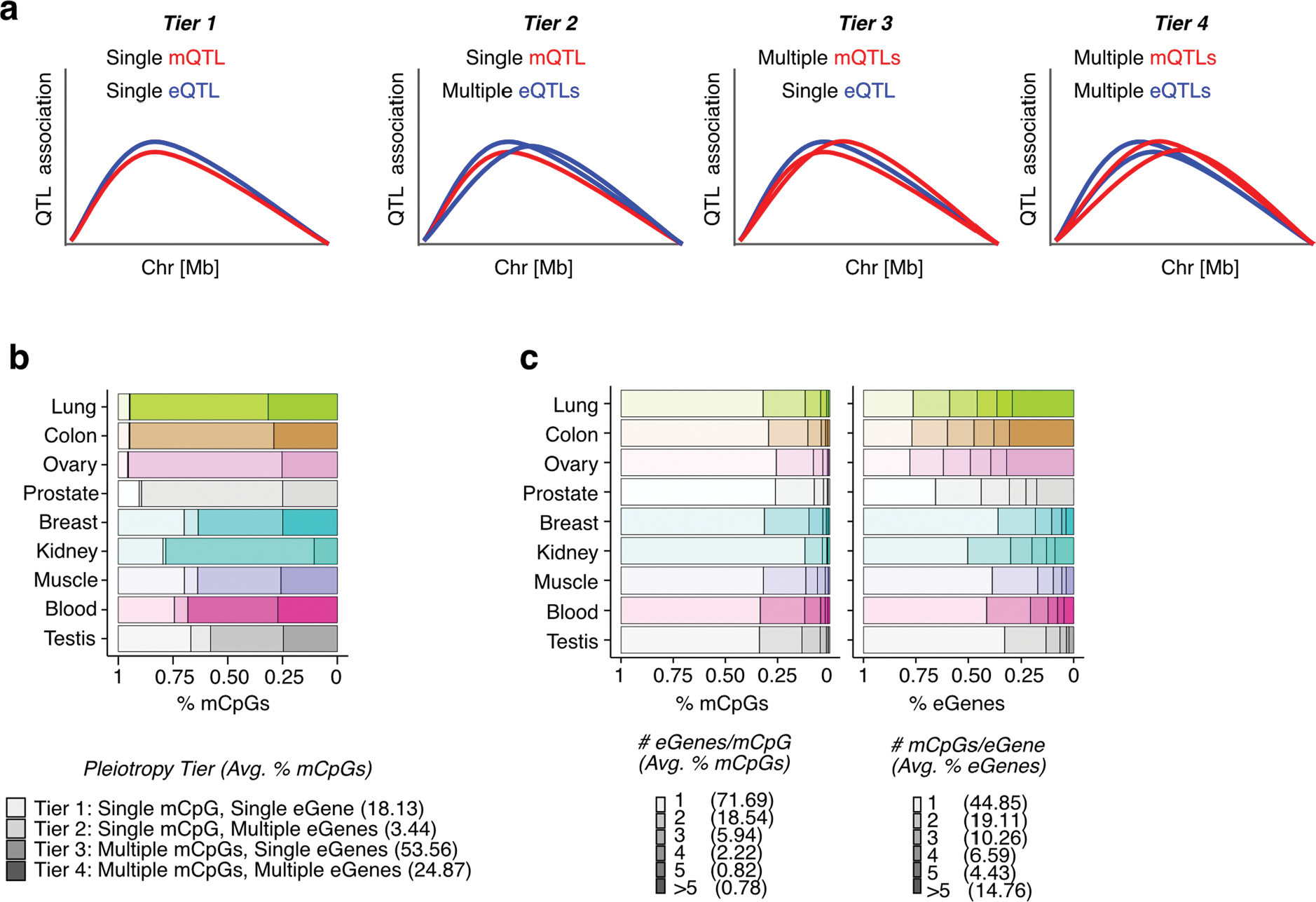
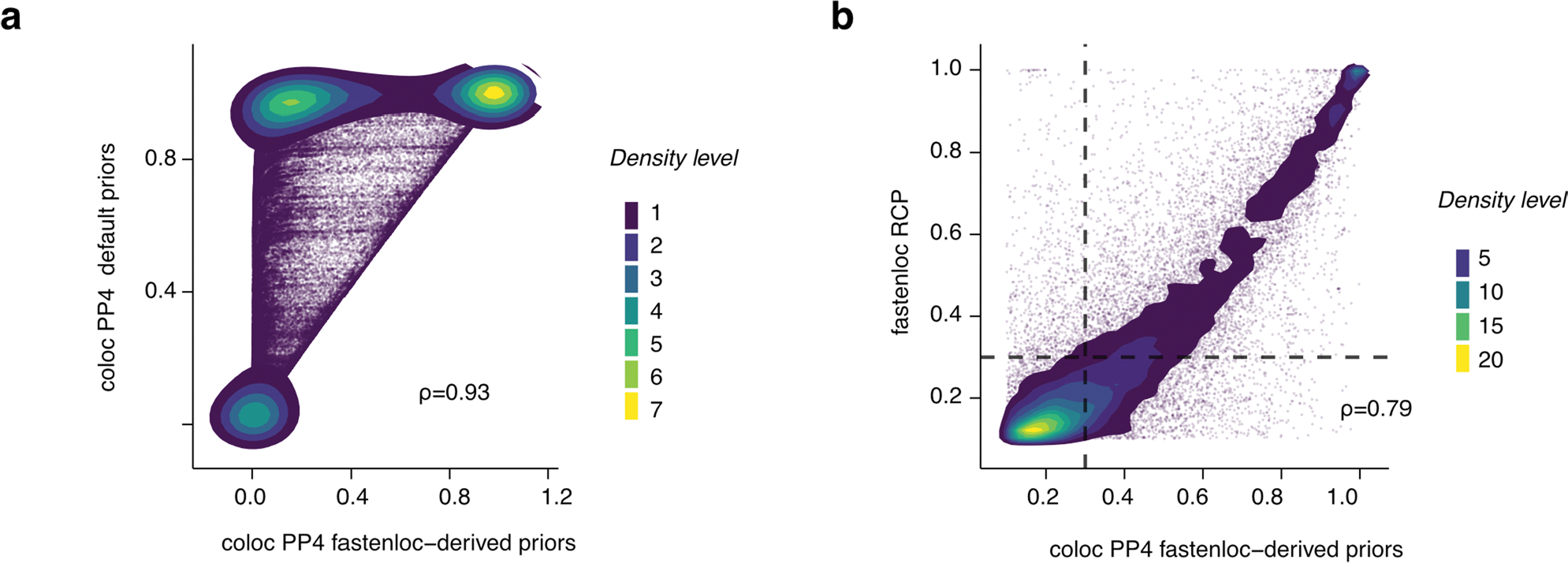
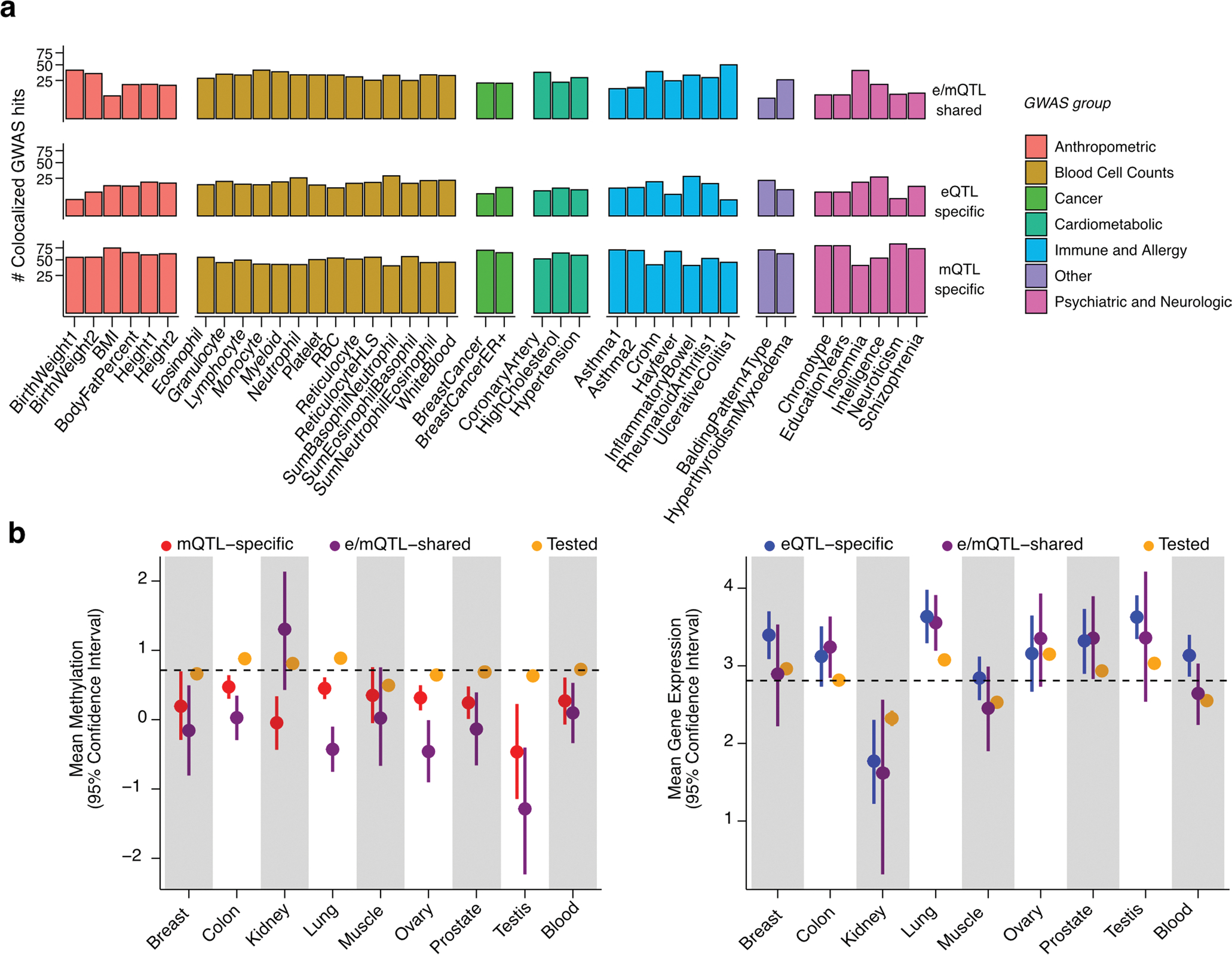
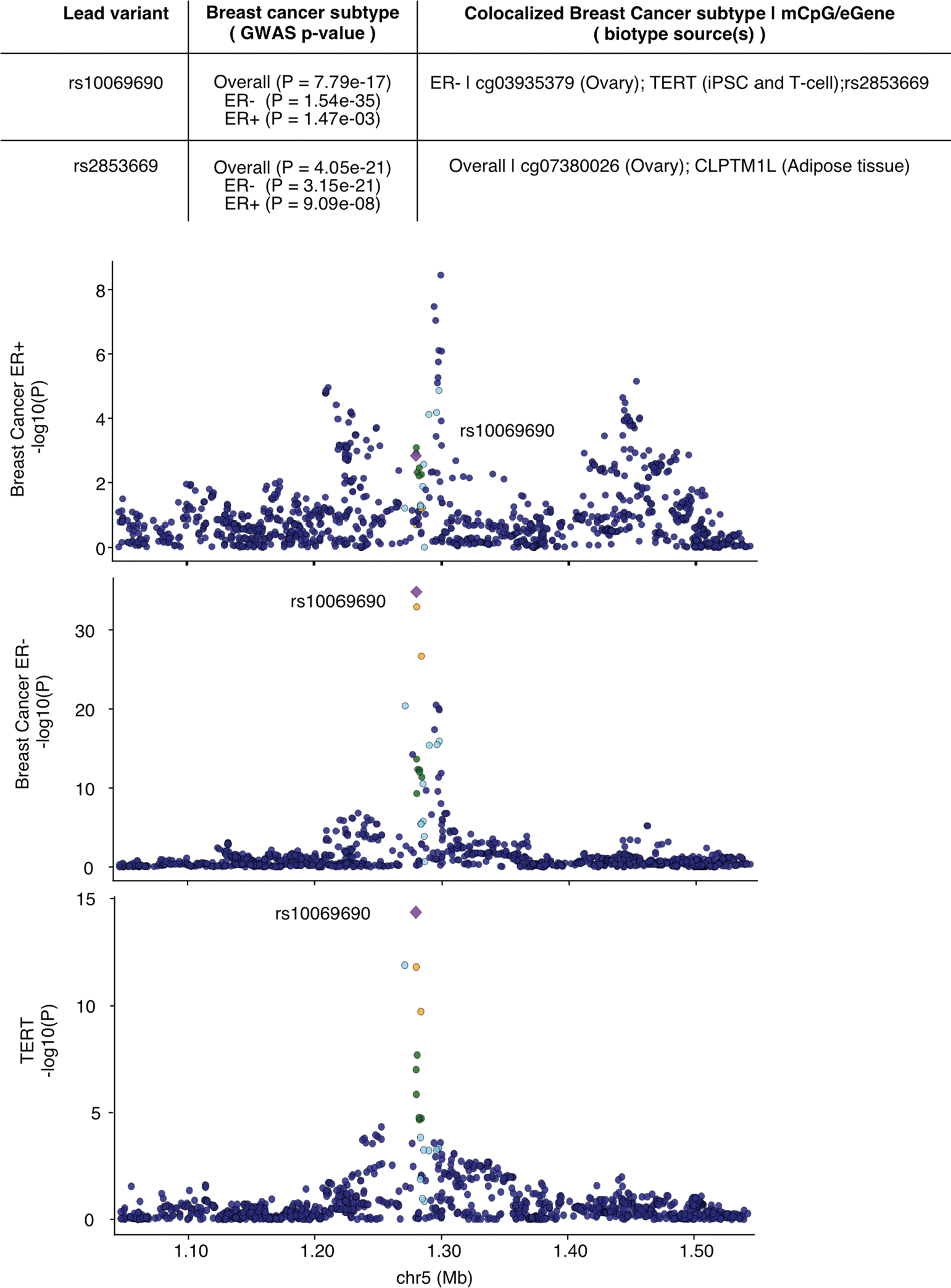
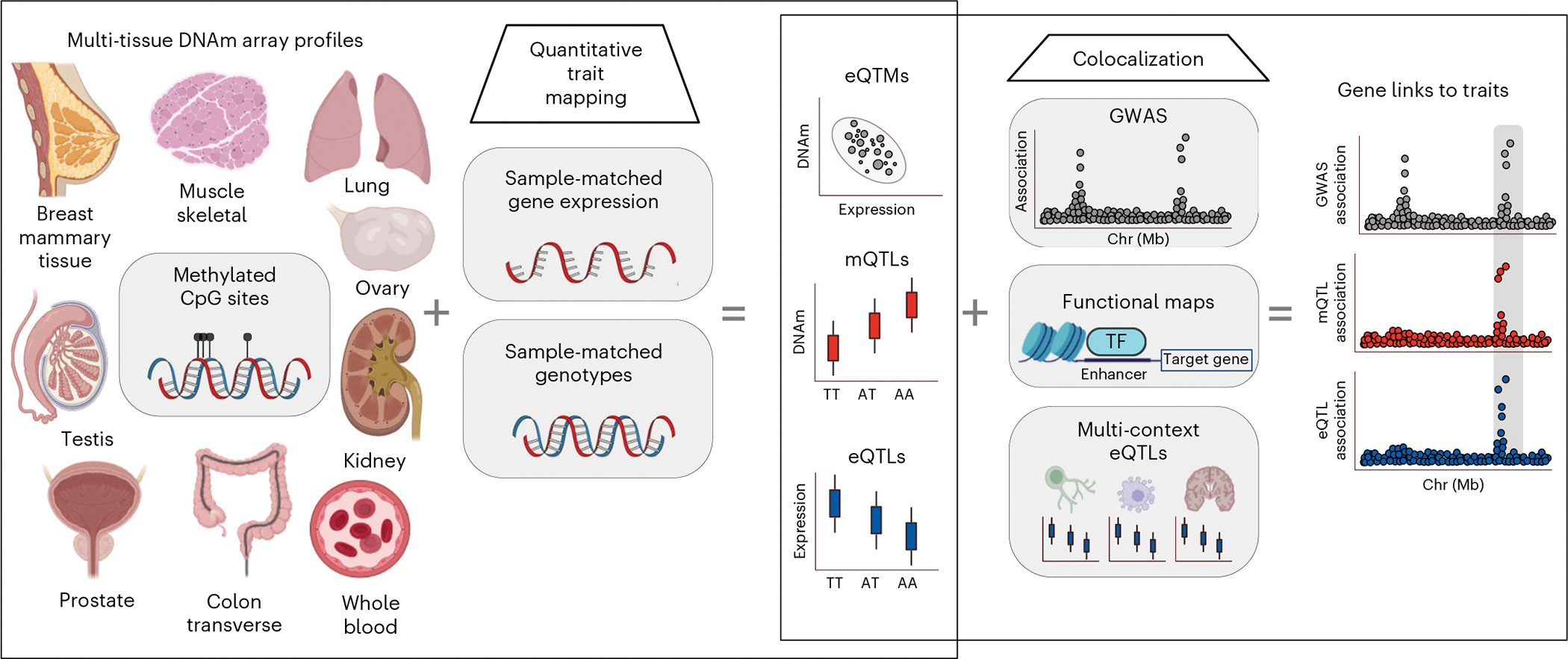
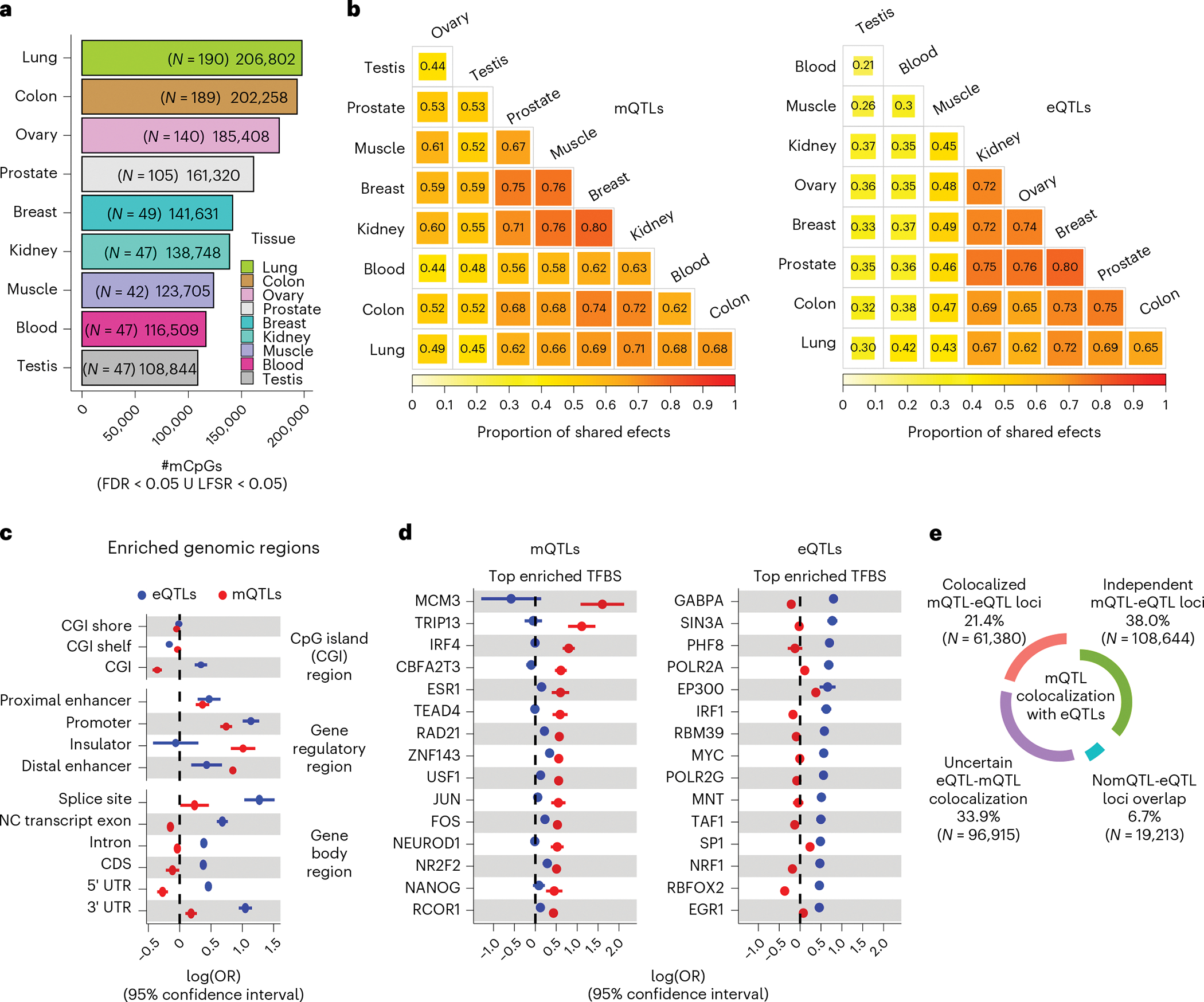
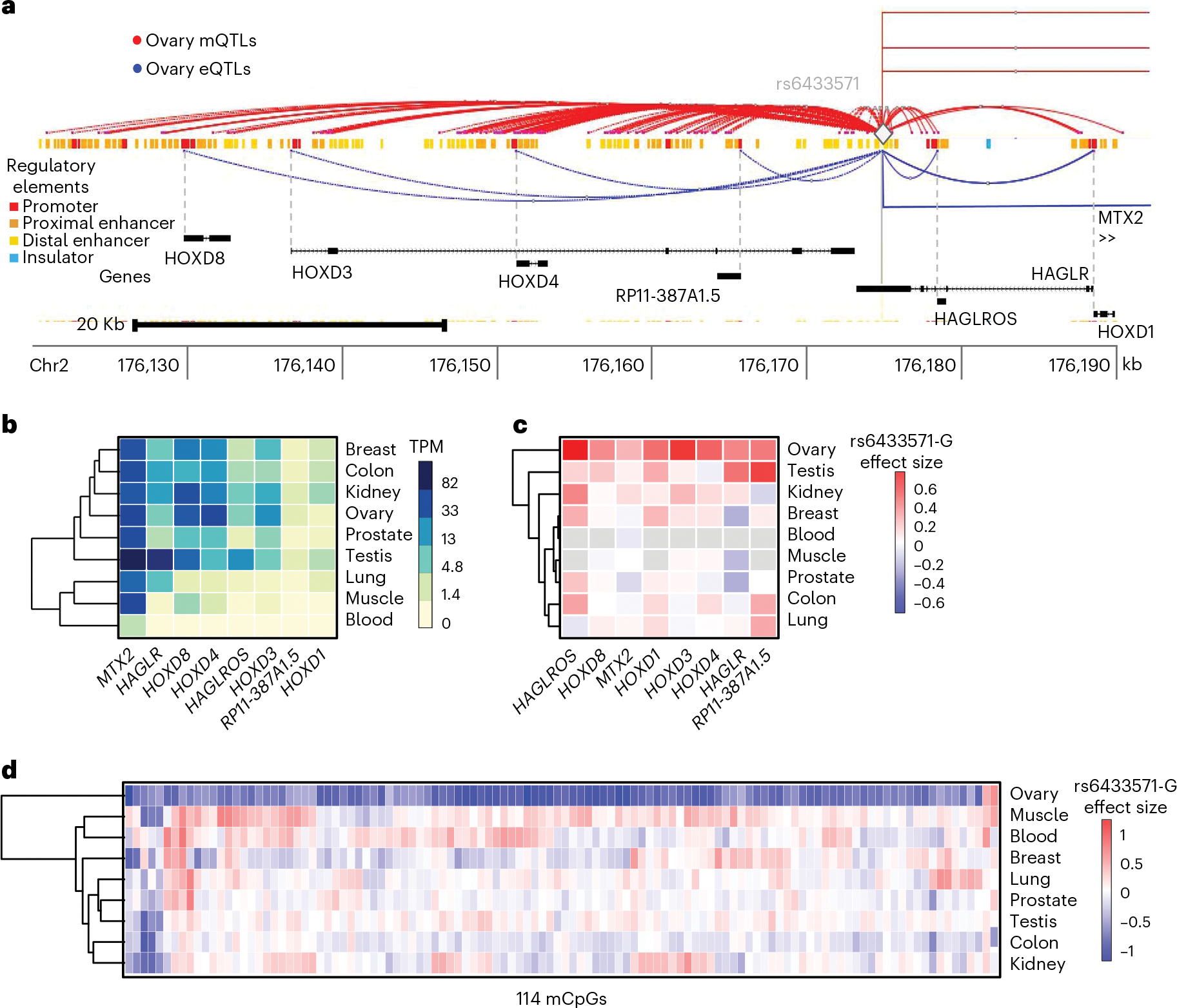
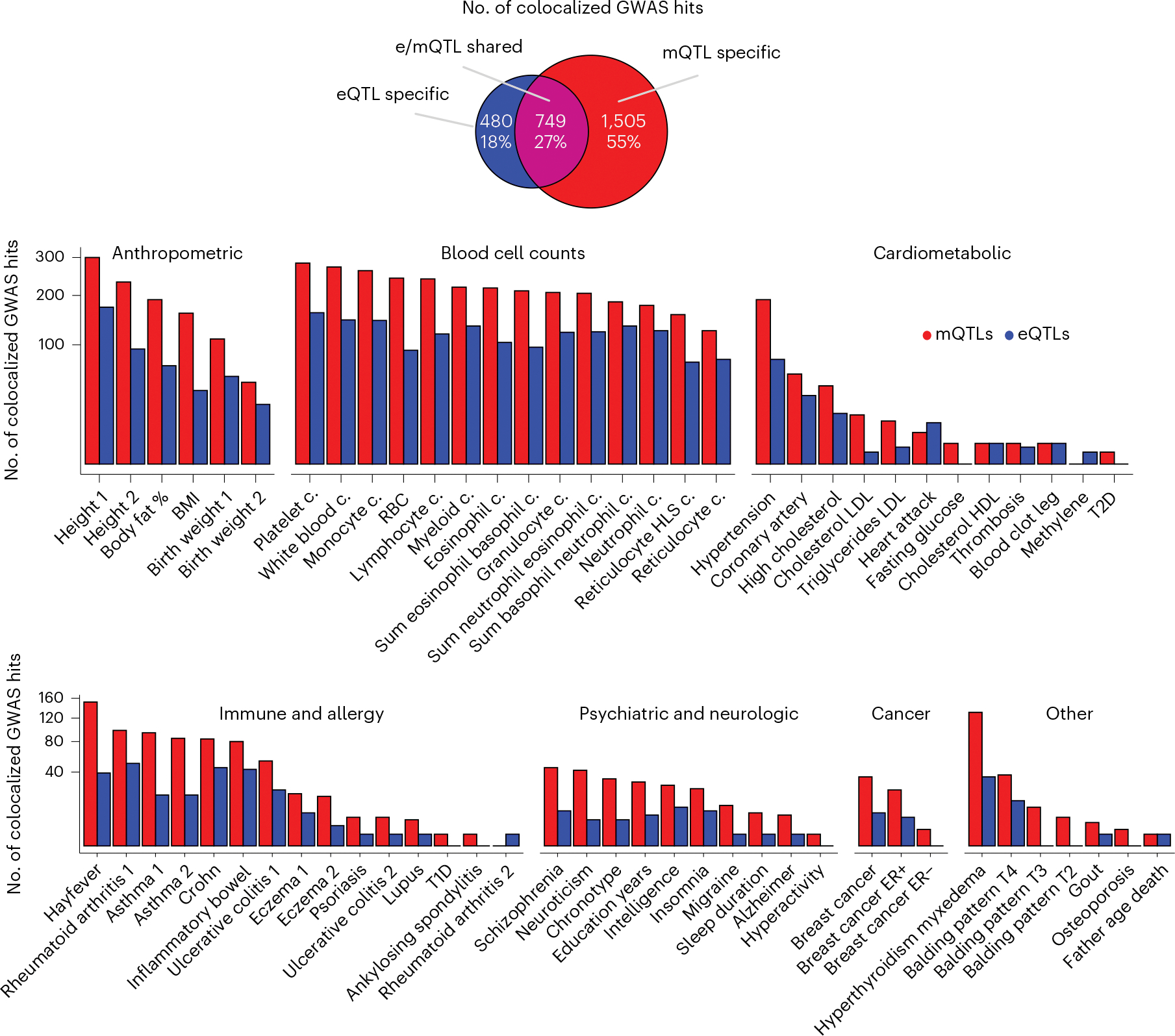
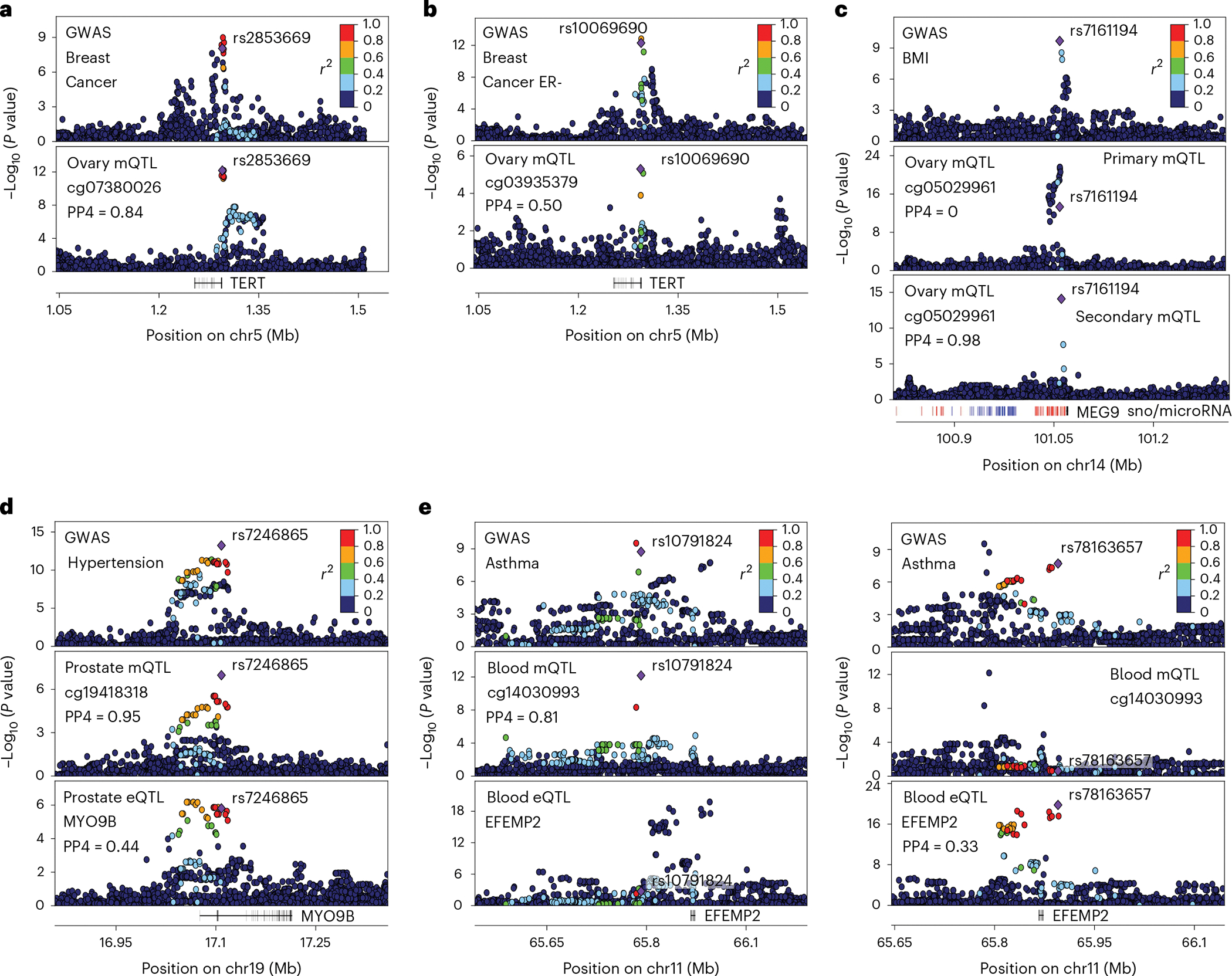
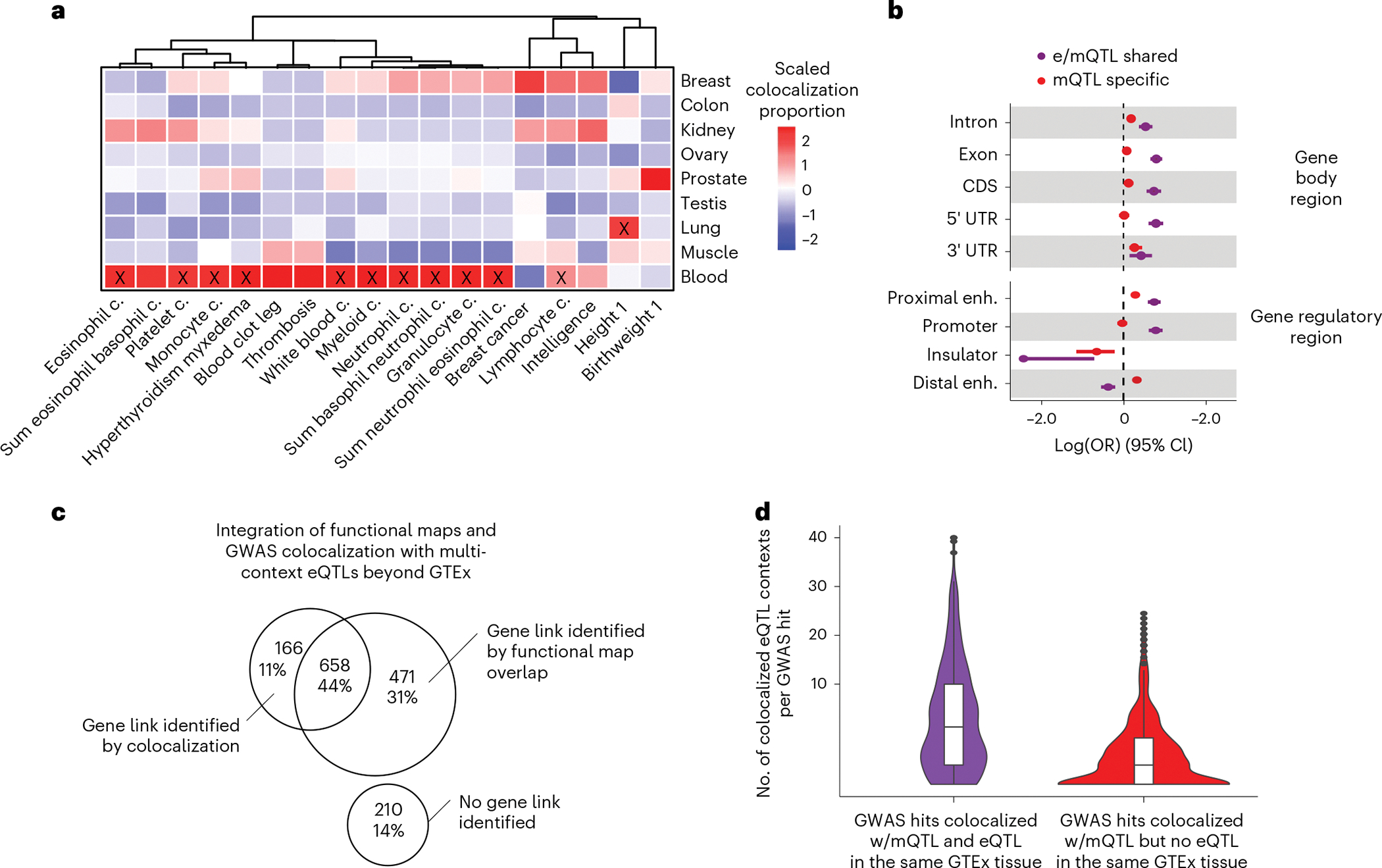
Studies of DNA methylation (DNAm) in solid human tissues are relatively scarce; tissue-specific characterization of DNAm is needed to understand its role in gene regulation and its relevance to complex traits. We generated array-based DNAm profiles for 987 human samples from the Genotype-Tissue Expression (GTEx) project, representing 9 tissue types and 424 subjects. We characterized methylome and transcriptome correlations (eQTMs), genetic regulation in cis (mQTLs and eQTLs) across tissues and e/mQTLs links to complex traits. We identified mQTLs for 286,152 CpG sites, many of which (>5%) show tissue specificity, and mQTL colocalizations with 2,254 distinct GWAS hits across 83 traits. For 91% of these loci, a candidate gene link was identified by integration of functional maps, including eQTMs, and/or eQTL colocalization, but only 33% of loci involved an eQTL and mQTL present in the same tissue type. With this DNAm-focused integrative analysis, we contribute to the understanding of molecular regulatory mechanisms in human tissues and their impact on complex traits.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
Comment in
-
DNA methylation provides molecular links underlying complex traits.Nat Genet. 2023 Jan;55(1):12-13. doi: 10.1038/s41588-022-01249-y. Nat Genet. 2023. PMID: 36593303 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
