

Mendelian randomization and genetic colocalization infer the effects of the multi-tissue proteome on 211 complex disease-related phenotypes
- PMID: 36510323
- PMCID: PMC9746220
- DOI: 10.1186/s13073-022-01140-9
Mendelian randomization and genetic colocalization infer the effects of the multi-tissue proteome on 211 complex disease-related phenotypes
Abstract
Background: Human proteins are widely used as drug targets. Integration of large-scale protein-level genome-wide association studies (GWAS) and disease-related GWAS has thus connected genetic variation to disease mechanisms via protein. Previous proteome-by-phenome-wide Mendelian randomization (MR) studies have been mainly focused on plasma proteomes. Previous MR studies using the brain proteome only reported protein effects on a set of pre-selected tissue-specific diseases. No studies, however, have used high-throughput proteomics from multiple tissues to perform MR on hundreds of phenotypes.
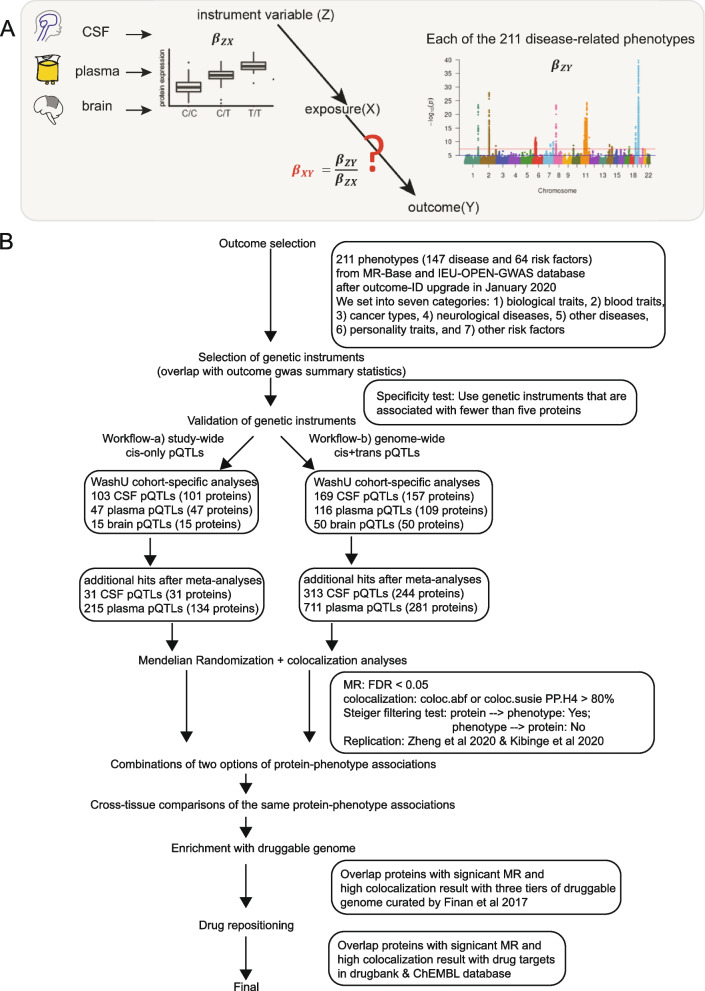
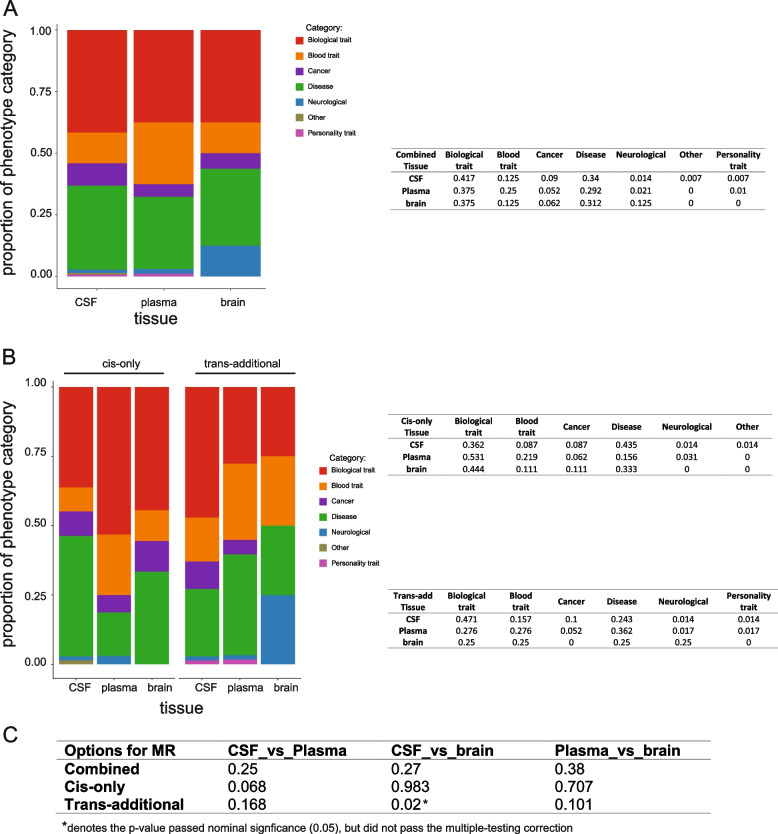
Methods: Here, we performed MR and colocalization analysis using multi-tissue (cerebrospinal fluid (CSF), plasma, and brain from pre- and post-meta-analysis of several disease-focus cohorts including Alzheimer disease (AD)) protein quantitative trait loci (pQTLs) as instrumental variables to infer protein effects on 211 phenotypes, covering seven broad categories: biological traits, blood traits, cancer types, neurological diseases, other diseases, personality traits, and other risk factors. We first implemented these analyses with cis pQTLs, as cis pQTLs are known for being less prone to horizontal pleiotropy. Next, we included both cis and trans conditionally independent pQTLs that passed the genome-wide significance threshold keeping only variants associated with fewer than five proteins to minimize pleiotropic effects. We compared the tissue-specific protein effects on phenotypes across different categories. Finally, we integrated the MR-prioritized proteins with the druggable genome to identify new potential targets.
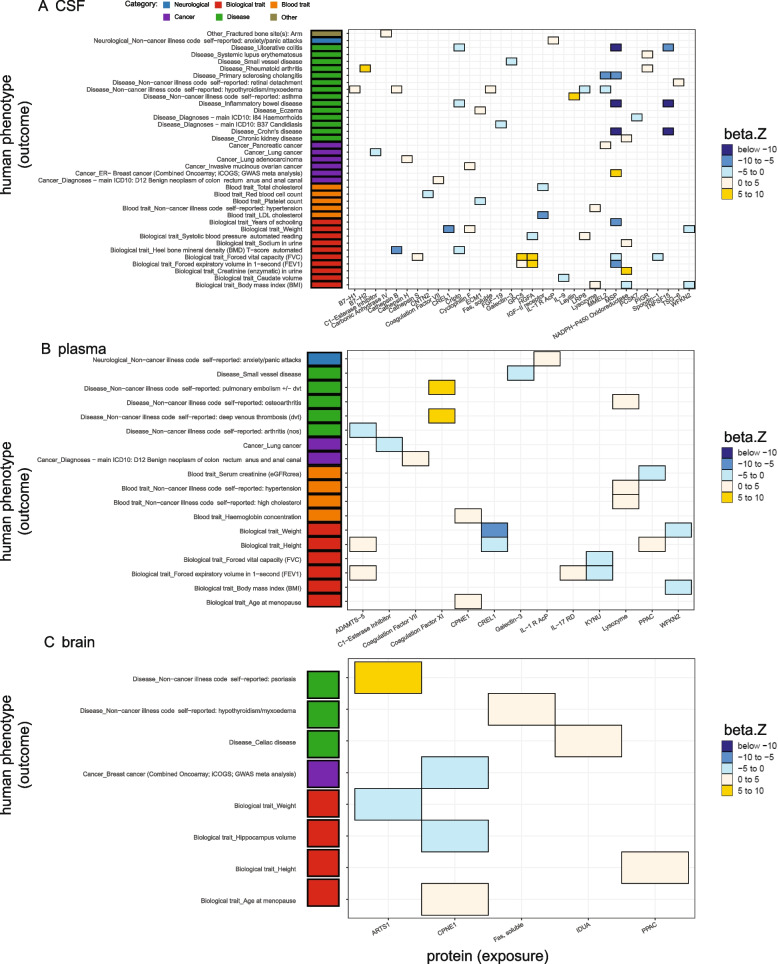
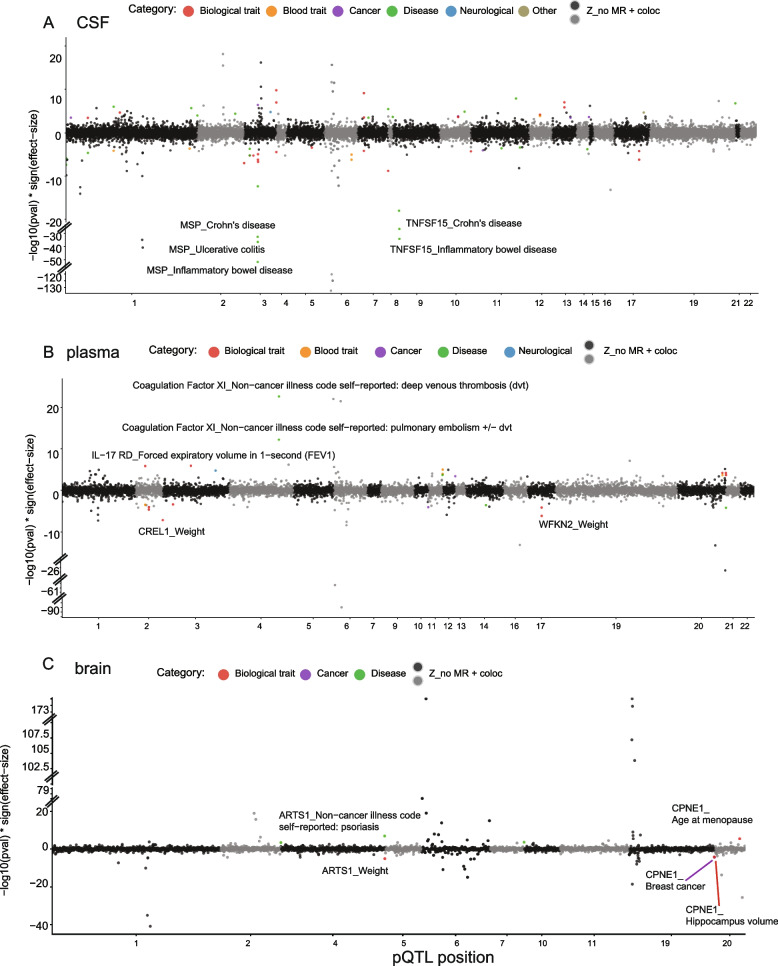
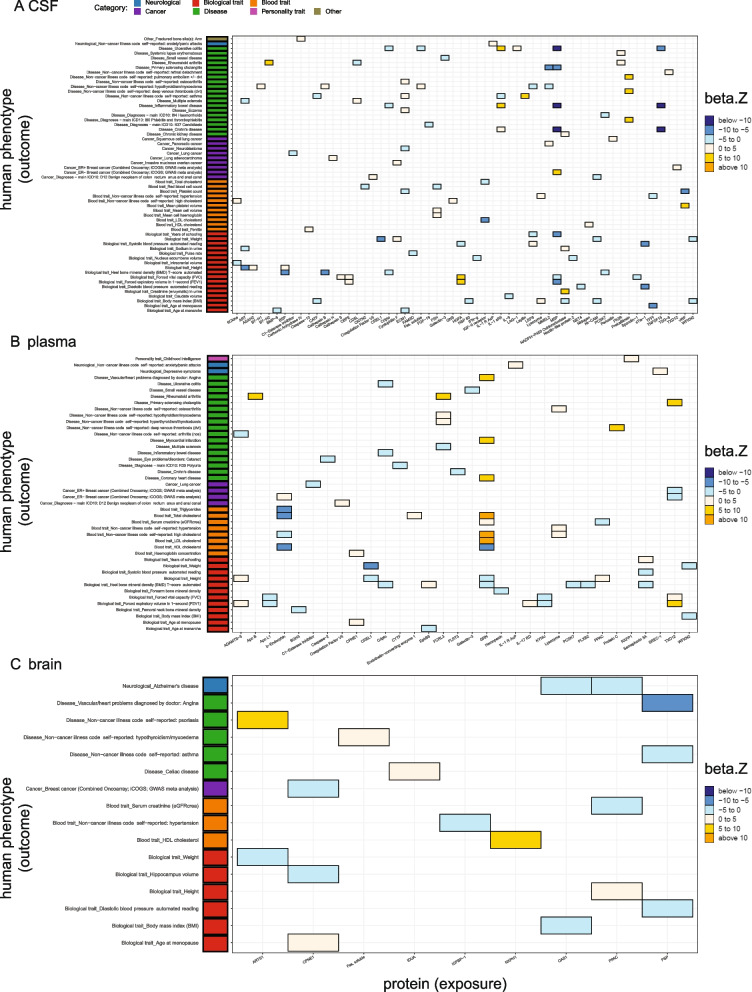
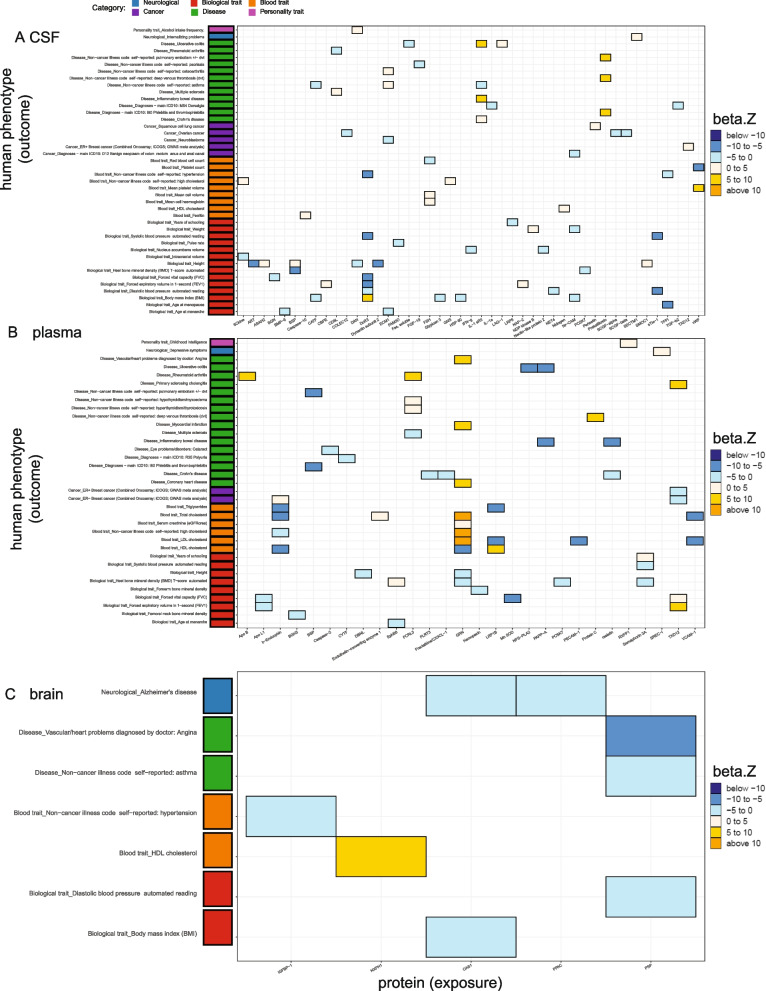
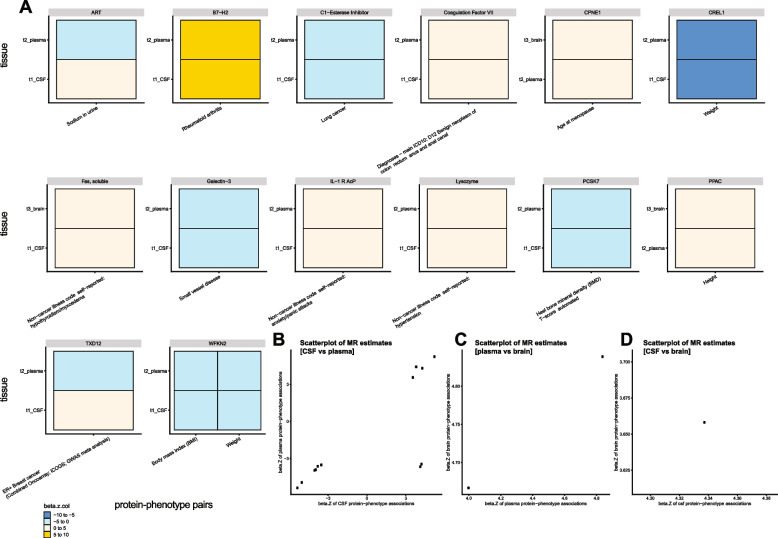
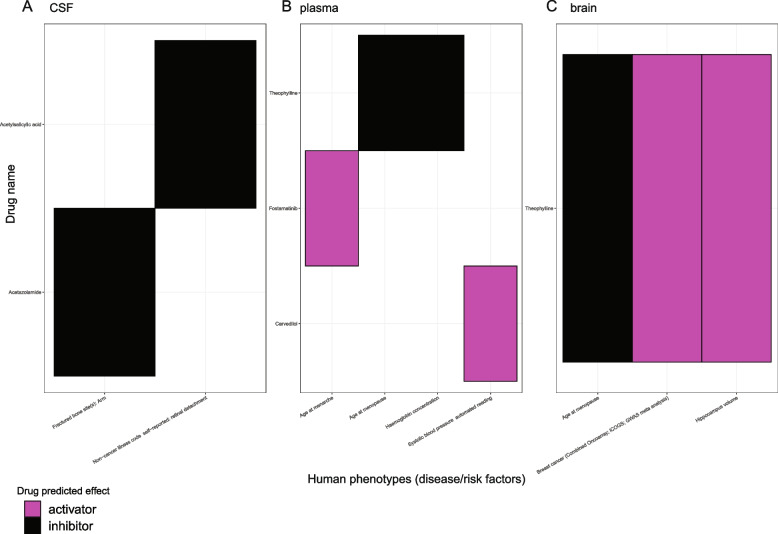
Results: In the MR and colocalization analysis including study-wide significant cis pQTLs as instrumental variables, we identified 33 CSF, 13 plasma, and five brain proteins to be putative causal for 37, 18, and eight phenotypes, respectively. After expanding the instrumental variables by including genome-wide significant cis and trans pQTLs, we identified a total of 58 CSF, 32 plasma, and nine brain proteins associated with 58, 44, and 16 phenotypes, respectively. For those protein-phenotype associations that were found in more than one tissue, the directions of the associations for 13 (87%) pairs were consistent across tissues. As we were unable to use methods correcting for horizontal pleiotropy given most of the proteins were only associated with one valid instrumental variable after clumping, we found that the observations of protein-phenotype associations were consistent with a causal role or horizontal pleiotropy. Between 66.7 and 86.3% of the disease-causing proteins overlapped with the druggable genome. Finally, between one and three proteins, depending on the tissue, were connected with at least one drug compound for one phenotype from both DrugBank and ChEMBL databases.
Conclusions: Integrating multi-tissue pQTLs with MR and the druggable genome may open doors to pinpoint novel interventions for complex traits with no effective treatments, such as ovarian and lung cancers.
Keywords: Complex human phenotypes; Genetic colocalization; Mendelian randomization; Multi-tissue proteomics; Protein quantitative trait loci.
© 2022. The Author(s).
Conflict of interest statement
CC receives research support from GSK. CC is a member of the advisory board of Vivid Genomics and Circular Genomics. The remaining authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
