

Structural variant analysis of a cancer reference cell line sample using multiple sequencing technologies
- PMID: 36514120
- PMCID: PMC9746098
- DOI: 10.1186/s13059-022-02816-6
Structural variant analysis of a cancer reference cell line sample using multiple sequencing technologies
Abstract
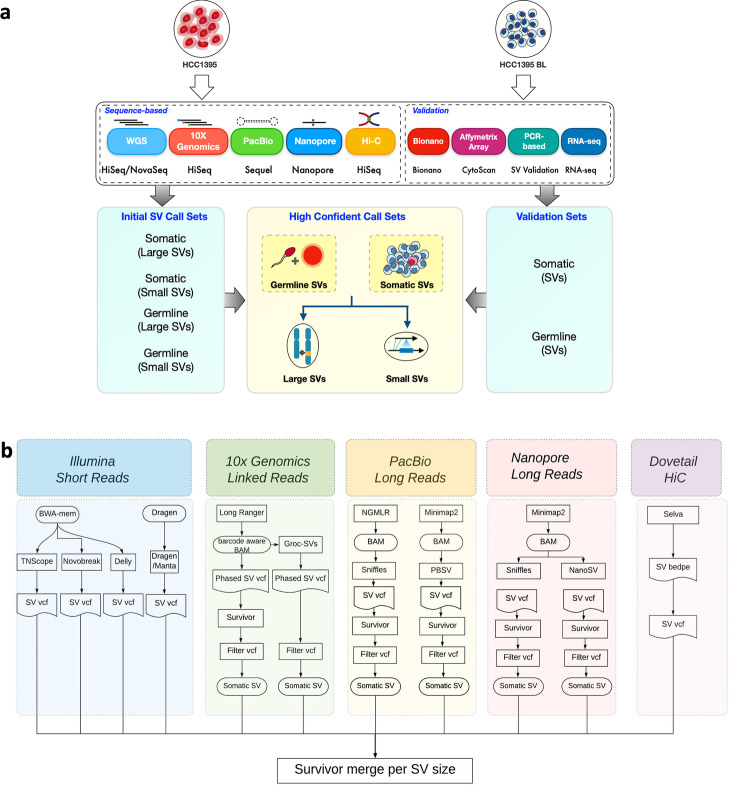
Background: The cancer genome is commonly altered with thousands of structural rearrangements including insertions, deletions, translocation, inversions, duplications, and copy number variations. Thus, structural variant (SV) characterization plays a paramount role in cancer target identification, oncology diagnostics, and personalized medicine. As part of the SEQC2 Consortium effort, the present study established and evaluated a consensus SV call set using a breast cancer reference cell line and matched normal control derived from the same donor, which were used in our companion benchmarking studies as reference samples.
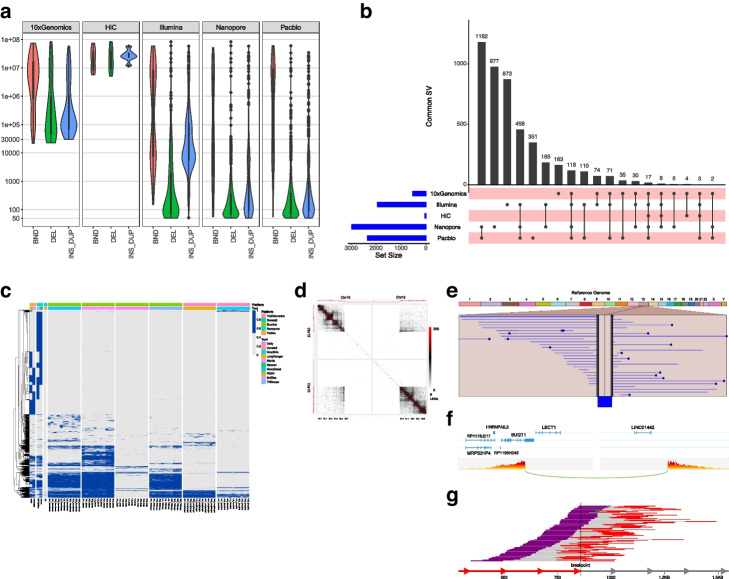
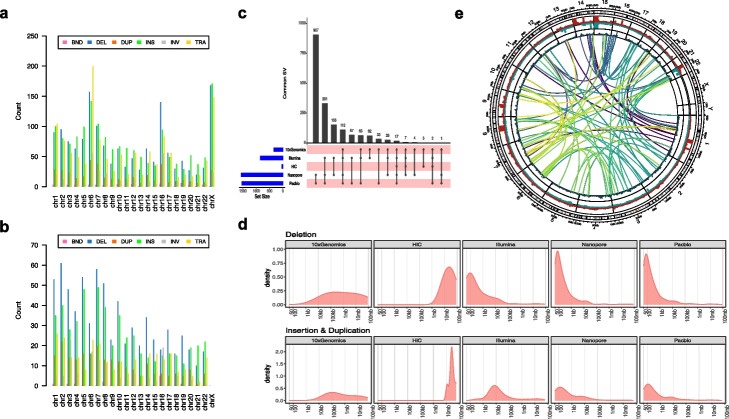
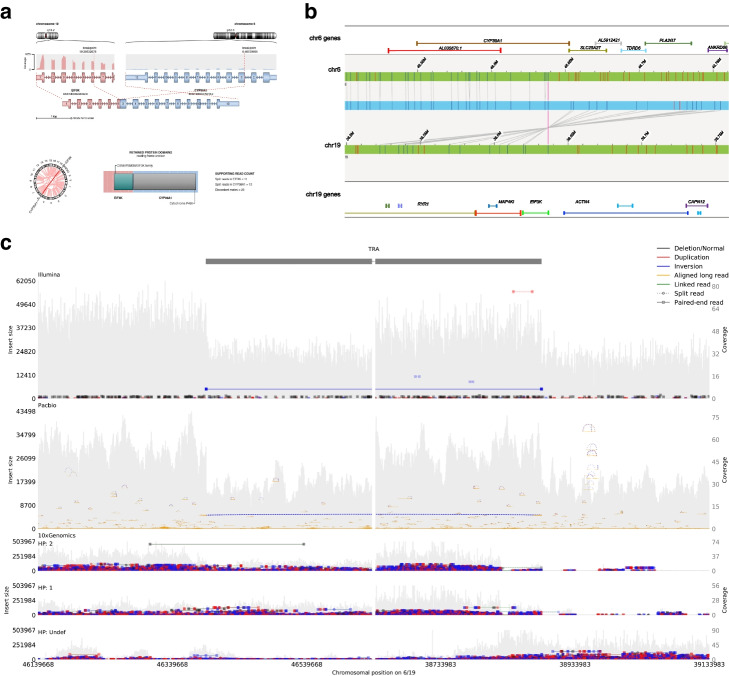
Results: We systematically investigated somatic SVs in the reference cancer cell line by comparing to a matched normal cell line using multiple NGS platforms including Illumina short-read, 10X Genomics linked reads, PacBio long reads, Oxford Nanopore long reads, and high-throughput chromosome conformation capture (Hi-C). We established a consensus SV call set of a total of 1788 SVs including 717 deletions, 230 duplications, 551 insertions, 133 inversions, 146 translocations, and 11 breakends for the reference cancer cell line. To independently evaluate and cross-validate the accuracy of our consensus SV call set, we used orthogonal methods including PCR-based validation, Affymetrix arrays, Bionano optical mapping, and identification of fusion genes detected from RNA-seq. We evaluated the strengths and weaknesses of each NGS technology for SV determination, and our findings provide an actionable guide to improve cancer genome SV detection sensitivity and accuracy.
Conclusions: A high-confidence consensus SV call set was established for the reference cancer cell line. A large subset of the variants identified was validated by multiple orthogonal methods.
Keywords: Cancer; Multiple platforms; Next-generation sequencing technology; Reference call set; Structural variant calling algorithm; Structural variation.
© 2022. The Author(s).
Conflict of interest statement
EJ, AN, AM, AG, TT, and RB are employees of Illumina Inc. ZL is currently an employee of Sentienon Inc. RK is an employee of Immuneering Corporation. MA. AP, BK, KH, and AH are employees of Bionano Genomics, and LTF is an employee of Roche Sequencing Solutions Inc. AA is an employee of DNAnexus. VM is an employee of Dovetail Genomics. All other authors declare that they have no competing interests.
Figures
References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Albertson DG, Ylstra B, Segraves R, Collins C, Dairkee SH, Kowbel D, et al. Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nat Genet. 2000;25:144. - PubMed
-
- Liggett WH, Sidransky D. Role of the p16 tumor suppressor gene in cancer. J Clin Oncol. 1998;16:1197–1206. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
