Fundamental Pathobiology of Coronary Atherosclerosis and Clinical Implications for Chronic Ischemic Heart Disease Management-The Plaque Hypothesis: A Narrative Review
- PMID: 36515941
- PMCID: PMC11016334
- DOI: 10.1001/jamacardio.2022.3926
Fundamental Pathobiology of Coronary Atherosclerosis and Clinical Implications for Chronic Ischemic Heart Disease Management-The Plaque Hypothesis: A Narrative Review
Abstract
Importance: Recent clinical and imaging studies underscore that major adverse cardiac events (MACE) outcomes are associated not solely with severe coronary obstructions (ischemia hypothesis or stenosis hypothesis), but with the plaque burden along the entire coronary tree. New research clarifies the pathobiologic mechanisms responsible for plaque development/progression/destabilization leading to MACE (plaque hypothesis), but the translation of these insights to clinical management strategies has lagged. This narrative review elaborates the plaque hypothesis and explicates the current understanding of underlying pathobiologic mechanisms, the provocative destabilizing influences, the diagnostic and therapeutic implications, and their actionable clinical management approaches to optimize the management of patients with chronic coronary disease.
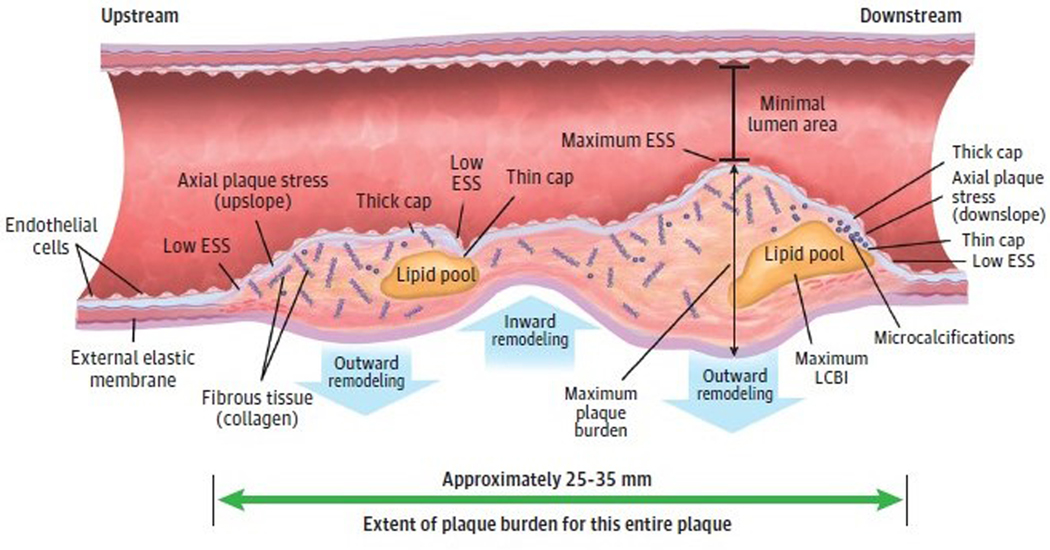
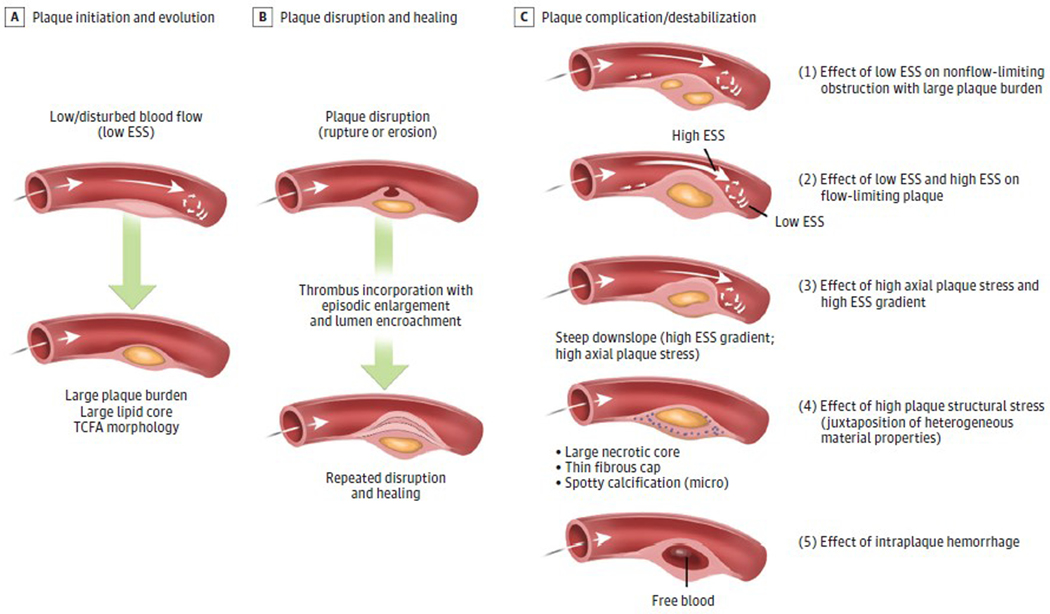
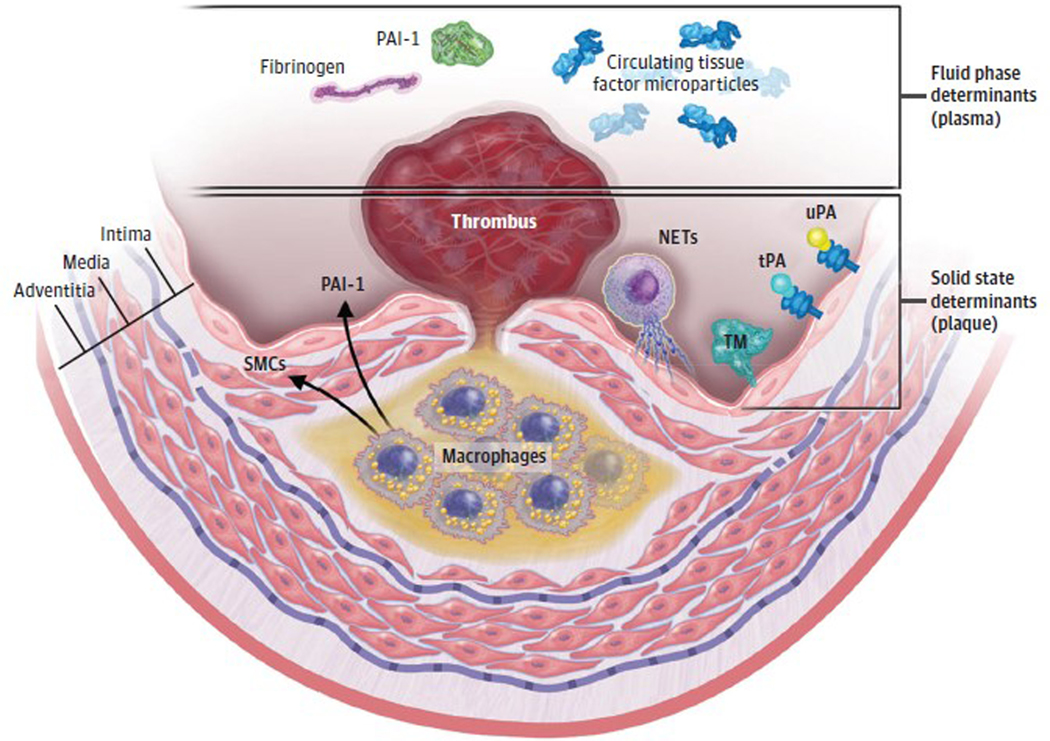
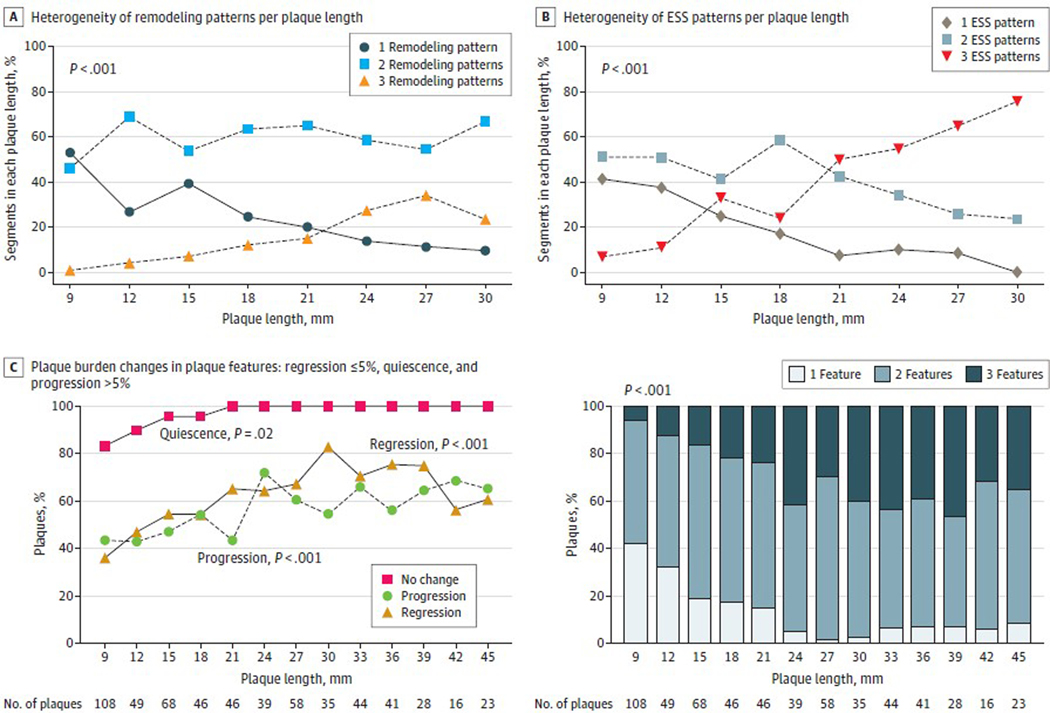
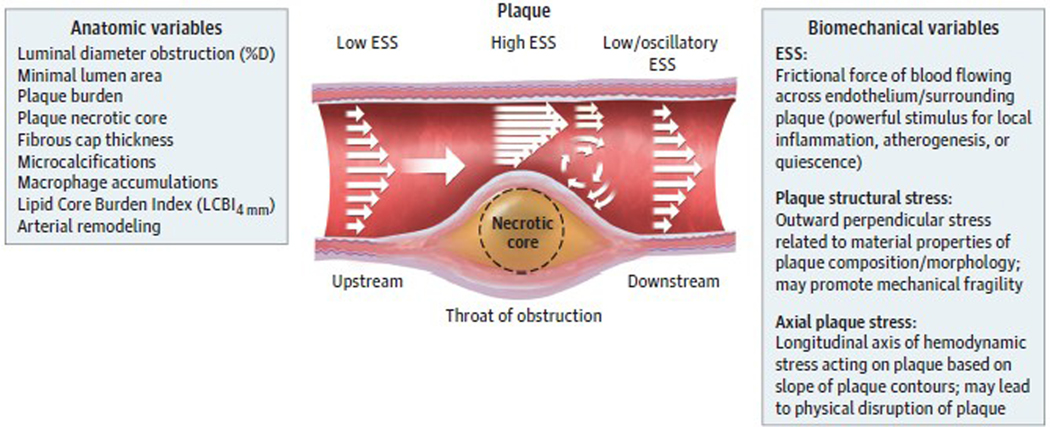
Observations: Clinical trials of management strategies for patients with chronic coronary artery disease demonstrate that while MACE rate increases progressively with the anatomic extent of coronary disease, revascularization of the ischemia-producing obstruction does not forestall MACE. Most severely obstructive coronary lesions often remain quiescent and seldom destabilize to cause a MACE. Coronary lesions that later provoke acute myocardial infarction often do not narrow the lumen critically. Invasive and noninvasive imaging can identify the plaque anatomic characteristics (plaque burden, plaque topography, lipid content) and local hemodynamic/biomechanical characteristics (endothelial shear stress, plaque structural stress, axial plaque stress) that can indicate the propensity of individual plaques to provoke a MACE.
Conclusions and relevance: The pathobiologic construct concerning the culprit region of a plaque most likely to cause a MACE (plaque hypothesis), which incorporates multiple convergent plaque features, informs the evolution of a new management strategy capable of identifying the high-risk portion of plaque wherever it is located along the course of the coronary artery. Ongoing investigations of high-risk plaque features, coupled with technical advances to enable prognostic characterization in real time and at the point of care, will soon enable evaluation of the entire length of the atheromatous coronary artery and broaden the target(s) of our therapeutic intervention to include all regions of the plaque (both flow limiting and nonflow limiting).
Conflict of interest statement
Figures