

Three assays for in-solution enrichment of ancient human DNA at more than a million SNPs
- PMID: 36517229
- PMCID: PMC9808625
- DOI: 10.1101/gr.276728.122
Three assays for in-solution enrichment of ancient human DNA at more than a million SNPs
Abstract
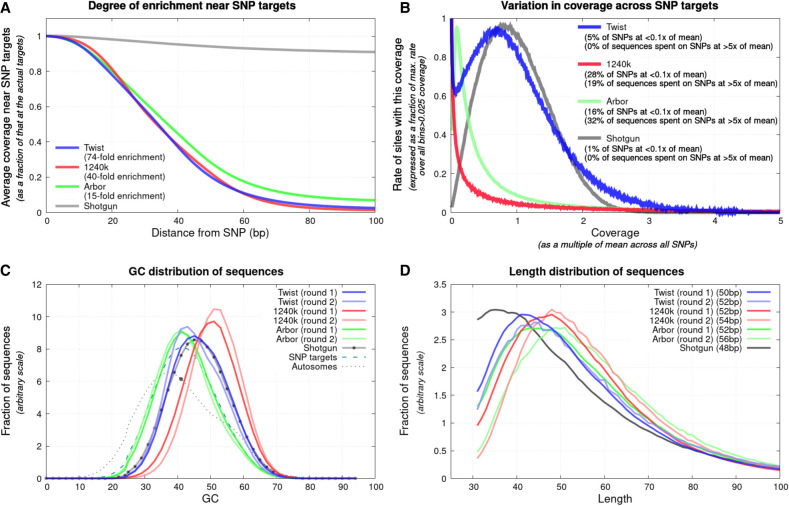
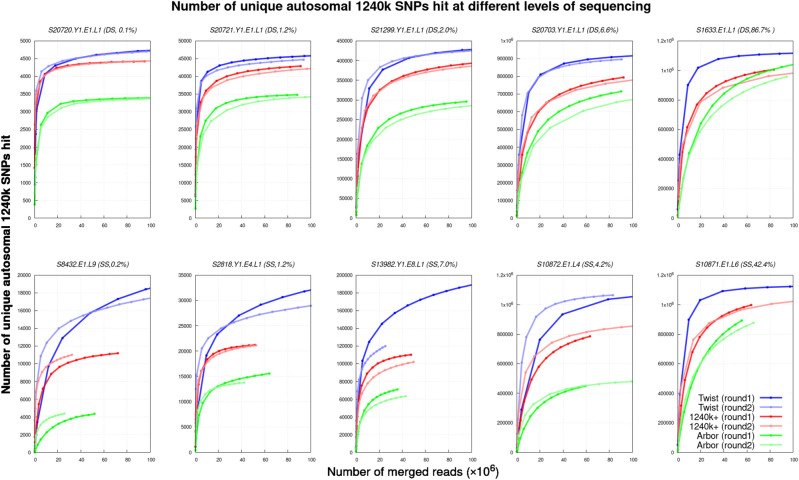
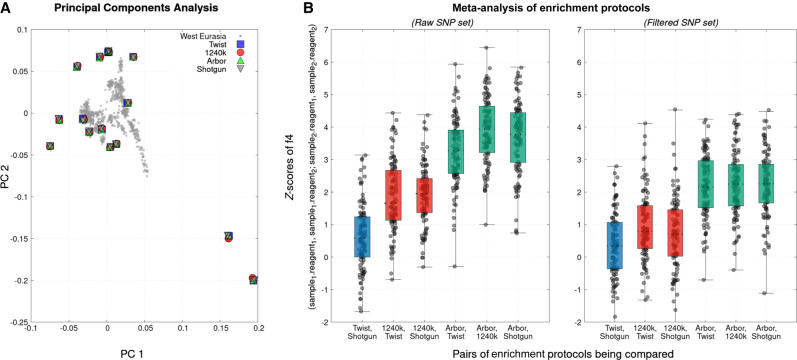
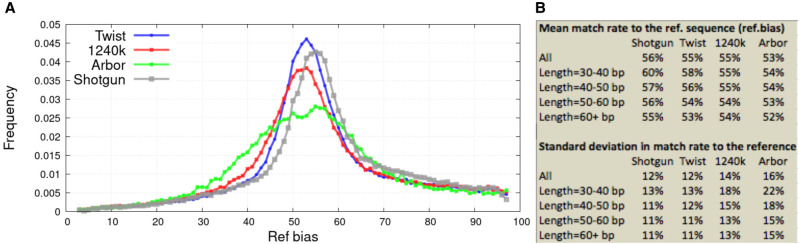
The strategy of in-solution enrichment for hundreds of thousands of single-nucleotide polymorphisms (SNPs) has been used to analyze >70% of individuals with genome-scale ancient DNA published to date. This approach makes it economical to study ancient samples with low proportions of human DNA and increases the rate of conversion of sampled remains into interpretable data. So far, nearly all such data have been generated using a set of bait sequences targeting about 1.24 million SNPs (the "1240k reagent"), but synthesis of the reagent has been cost-effective for only a few laboratories. In 2021, two companies, Daicel Arbor Biosciences and Twist Bioscience, made available assays that target the same core set of SNPs along with supplementary content. We test all three assays on a common set of 27 ancient DNA libraries and show that all three are effective at enriching many hundreds of thousands of SNPs. For all assays, one round of enrichment produces data that are as useful as two. In our testing, the "Twist Ancient DNA" assay produces the highest coverages, greatest uniformity on targeted positions, and almost no bias toward enriching one allele more than another relative to shotgun sequencing. We also identify hundreds of thousands of targeted SNPs for which there is minimal allelic bias when comparing 1240k data to either shotgun or Twist data. This facilitates coanalysis of the large data sets that have been generated using 1240k and Twist capture, as well as shotgun sequencing approaches.
© 2022 Rohland et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Carpenter ML, Buenrostro JD, Valdiosera C, Schroeder H, Allentoft ME, Sikora M, Rasmussen M, Gravel S, Guillén S, Nekhrizov G, et al. 2013. Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am J Hum Genet 93: 852–864. 10.1016/j.ajhg.2013.10.002 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources