

Ovarian cancer mutational processes drive site-specific immune evasion
- PMID: 36517593
- PMCID: PMC9771812
- DOI: 10.1038/s41586-022-05496-1
Ovarian cancer mutational processes drive site-specific immune evasion
Abstract
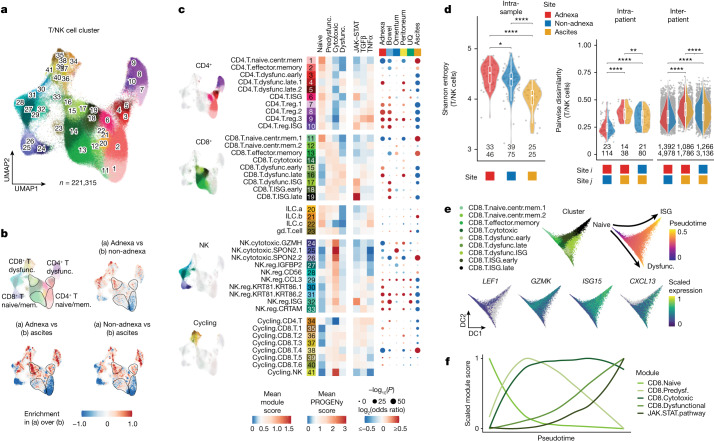
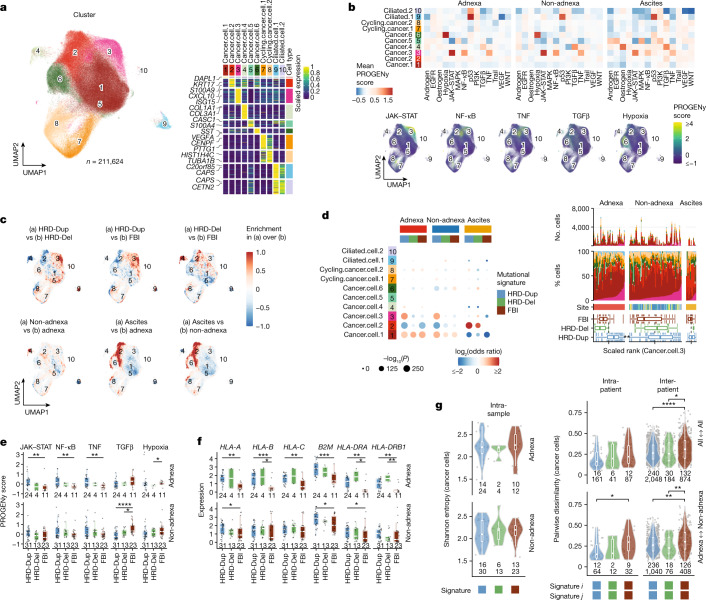
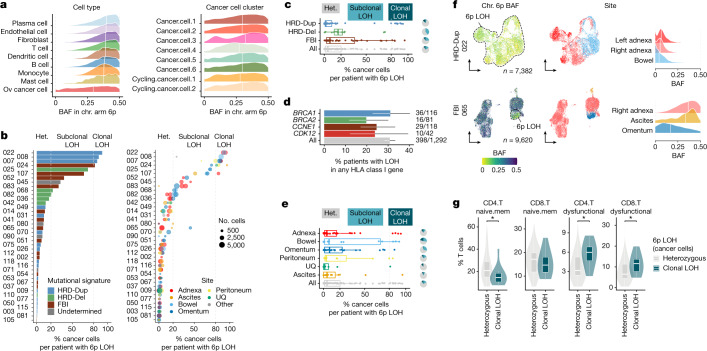
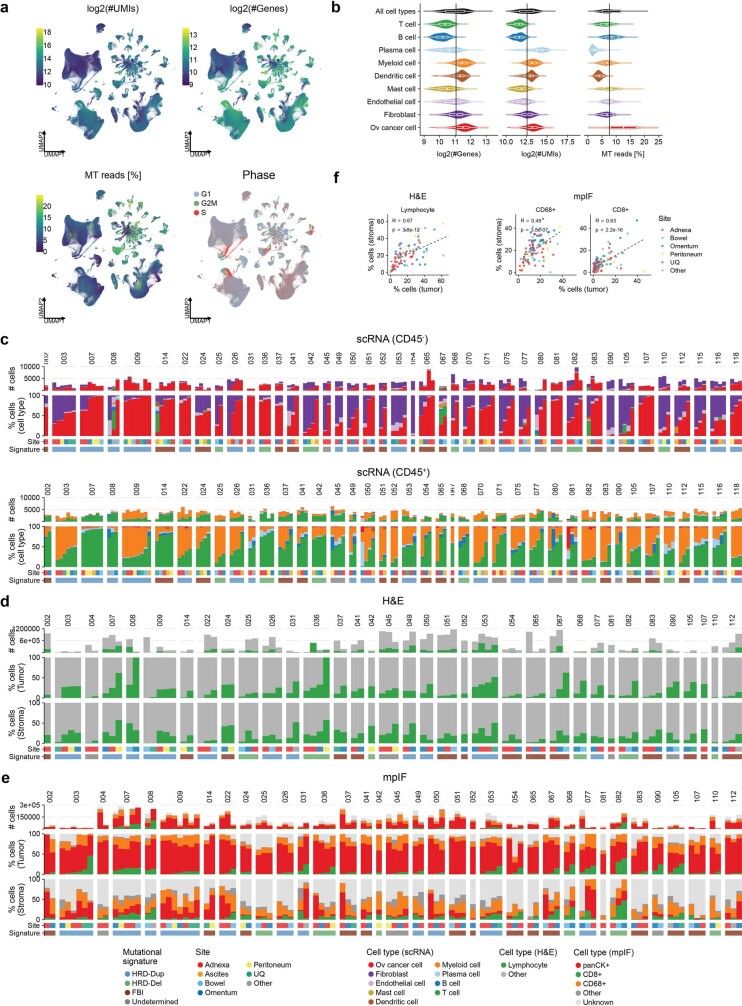
High-grade serous ovarian cancer (HGSOC) is an archetypal cancer of genomic instability1-4 patterned by distinct mutational processes5,6, tumour heterogeneity7-9 and intraperitoneal spread7,8,10. Immunotherapies have had limited efficacy in HGSOC11-13, highlighting an unmet need to assess how mutational processes and the anatomical sites of tumour foci determine the immunological states of the tumour microenvironment. Here we carried out an integrative analysis of whole-genome sequencing, single-cell RNA sequencing, digital histopathology and multiplexed immunofluorescence of 160 tumour sites from 42 treatment-naive patients with HGSOC. Homologous recombination-deficient HRD-Dup (BRCA1 mutant-like) and HRD-Del (BRCA2 mutant-like) tumours harboured inflammatory signalling and ongoing immunoediting, reflected in loss of HLA diversity and tumour infiltration with highly differentiated dysfunctional CD8+ T cells. By contrast, foldback-inversion-bearing tumours exhibited elevated immunosuppressive TGFβ signalling and immune exclusion, with predominantly naive/stem-like and memory T cells. Phenotypic state associations were specific to anatomical sites, highlighting compositional, topological and functional differences between adnexal tumours and distal peritoneal foci. Our findings implicate anatomical sites and mutational processes as determinants of evolutionary phenotypic divergence and immune resistance mechanisms in HGSOC. Our study provides a multi-omic cellular phenotype data substrate from which to develop and interpret future personalized immunotherapeutic approaches and early detection research.
© 2022. The Author(s).
Conflict of interest statement
S.P.S. is a shareholder of Imagia Canexia Health and a consultant to AstraZeneca, outside the scope of this study. D.Z. reports research funding to MSK from AstraZeneca, Genentech, Synthekine and Plexxikon; personal fees from Synlogic Therapeutics, Hookipa Biotech, Agenus, Synthekine, Memgen, Mana Therapeutics, Tessa Therapeutics and Xencor; and stock options from Accurius, Calidi Biotherapeutics and Immunos. D.Z. is an inventor on a patent concerning the use of Newcastle disease virus as a cancer therapeutic, licensed to Merck. C.F.F. reports research funding to the institution from Merck, AstraZeneca, Genentech/Roche, Bristol Myers Squibb and Daiichi and uncompensated membership of a scientific advisory board for Merck and Genentech and is a consultant for OncLive, Aptitude Health, Bristol Myers Squibb and Seagen, all outside the scope of this manuscript. B.W. reports ad hoc membership of the scientific advisory board of Repare Therapeutics, outside the scope of the submitted work. C.A. reports grants from Clovis, Genentech, AbbVie and AstraZeneca and personal fees from Tesaro, Eisai/Merck, Mersana Therapeutics, Roche/Genentech, Abbvie, AstraZeneca/Merck and Repare Therapeutics, outside the scope of the submitted work. N.R.A.-R. reports grants to MSK from Stryker/Novadaq and GRAIL, outside the scope of the submitted work. D.S.C. is on the medical advisory board of Apyx Medical Co, Verthermia Acquio and Biom'up and is a stockholder of Intuitive Surgical and TransEnterix. R.N.G. reports funding from GSK, Novartis, Mateon Therapeutics, Corcept, Regeneron, Clovis, Context Therapeutics, EMD Serono, MCM Education, OncLive, Aptitude Health and Prime Oncology, outside the scope of this work. Y.L.L. reports research funding from AstraZeneca, GSK/Tesaro and Tesaro Therapeutics outside the scope of this work. Y.L. consults for Calyx Clinical Trials Solutions and holds shares of Y-mAbs Therapeutics. T.J.H. receives research funding from Bristol Myers Squibb, Calico Labs and the Parker Institute for Cancer Immunotherapy. S.F.B. owns equity in, receives compensation from, and serves as a consultant and on the scientific advisory board and board of directors of Volastra Therapeutics. The other authors declare no competing interests.
Figures
















Comment in
-
Genetics and anatomy sculpt immune-cell partners of ovarian cancer.Nature. 2022 Dec;612(7941):634-636. doi: 10.1038/d41586-022-04169-3. Nature. 2022. PMID: 36517683 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
