

STRling: a k-mer counting approach that detects short tandem repeat expansions at known and novel loci
- PMID: 36517892
- PMCID: PMC9753380
- DOI: 10.1186/s13059-022-02826-4
STRling: a k-mer counting approach that detects short tandem repeat expansions at known and novel loci
Abstract
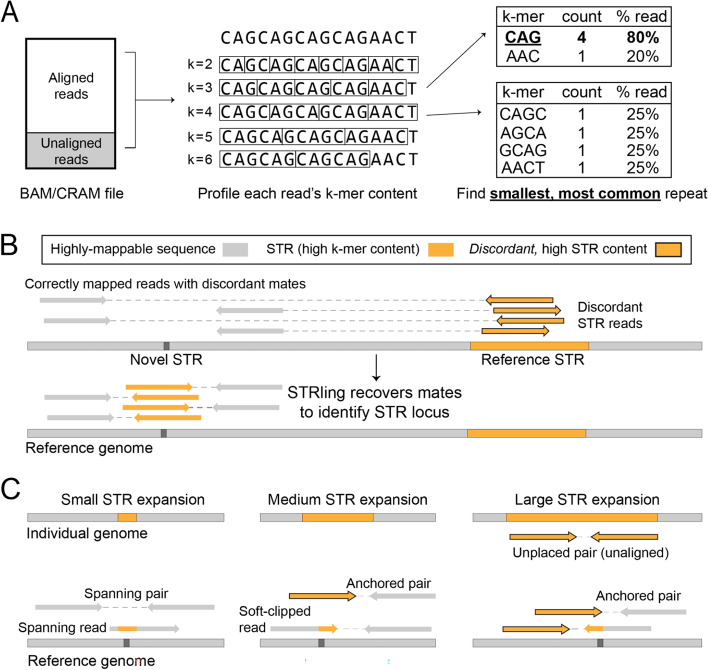
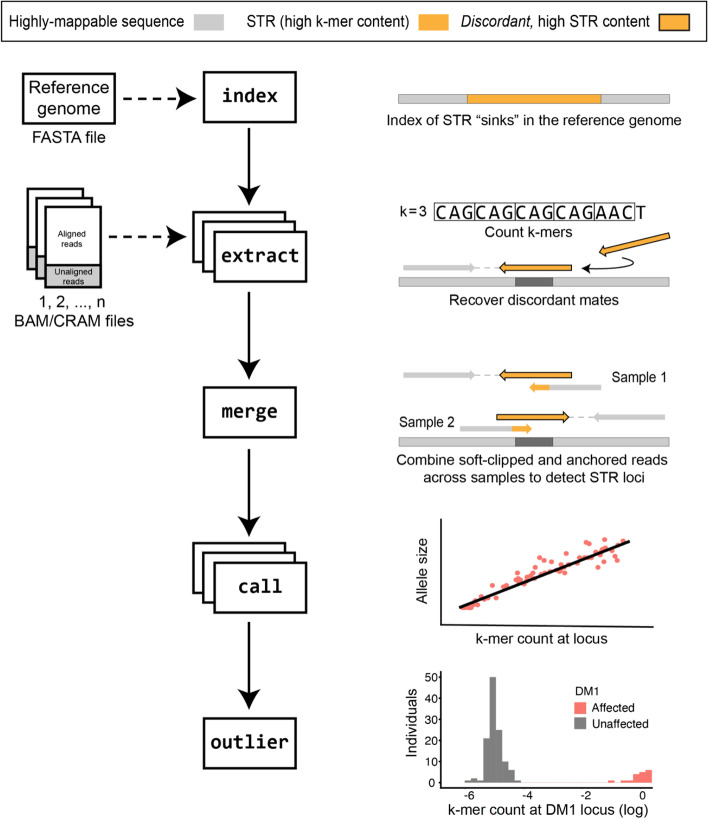
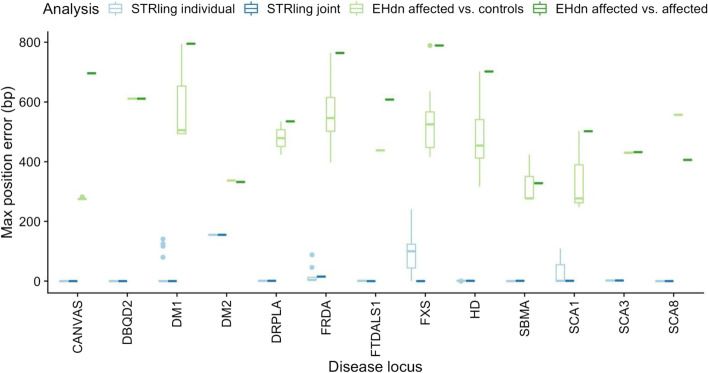
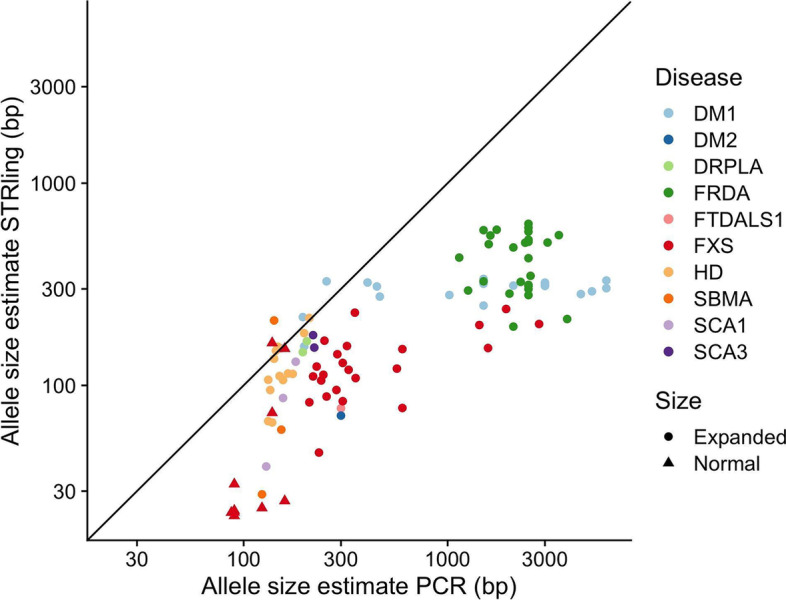
Expansions of short tandem repeats (STRs) cause many rare diseases. Expansion detection is challenging with short-read DNA sequencing data since supporting reads are often mapped incorrectly. Detection is particularly difficult for "novel" STRs, which include new motifs at known loci or STRs absent from the reference genome. We developed STRling to efficiently count k-mers to recover informative reads and call expansions at known and novel STR loci. STRling is sensitive to known STR disease loci, has a low false discovery rate, and resolves novel STR expansions to base-pair position accuracy. It is fast, scalable, open-source, and available at: github.com/quinlan-lab/STRling .
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
