

Restriction Endonuclease-Based Modification-Dependent Enrichment (REMoDE) of DNA for Metagenomic Sequencing
- PMID: 36519847
- PMCID: PMC9888230
- DOI: 10.1128/aem.01670-22
Restriction Endonuclease-Based Modification-Dependent Enrichment (REMoDE) of DNA for Metagenomic Sequencing
Abstract
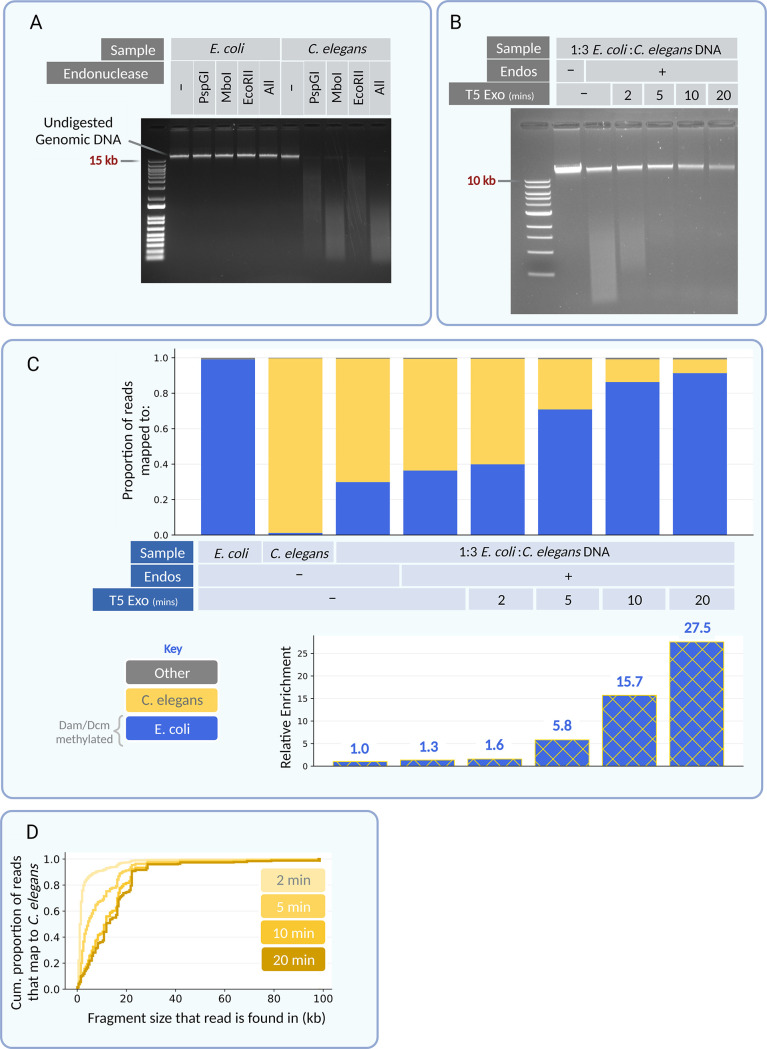
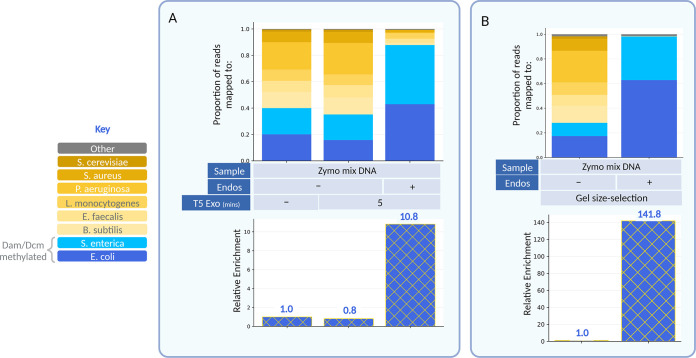
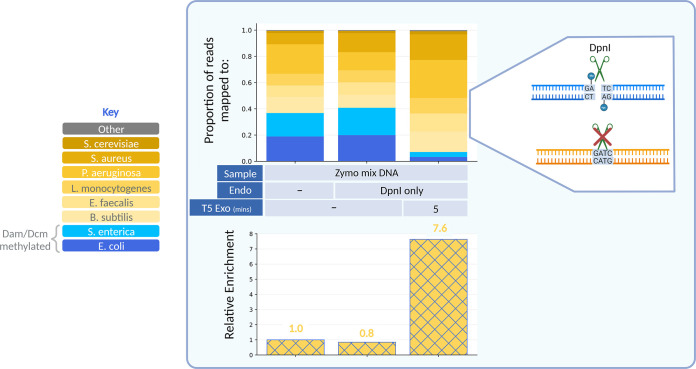
Metagenomic sequencing is a swift and powerful tool to ascertain the presence of an organism of interest in a sample. However, sequencing coverage of the organism of interest can be insufficient due to an inundation of reads from irrelevant organisms in the sample. Here, we report a nuclease-based approach to rapidly enrich for DNA from certain organisms, including enterobacteria, based on their differential endogenous modification patterns. We exploit the ability of taxon-specific methylated motifs to resist the action of cognate methylation-sensitive restriction endonucleases that thereby digest unwanted, unmethylated DNA. Subsequently, we use a distributive exonuclease or electrophoretic separation to deplete or exclude the digested fragments, thus enriching for undigested DNA from the organism of interest. As a proof of concept, we apply this method to enrich for the enterobacteria Escherichia coli and Salmonella enterica by 11- to 142-fold from mock metagenomic samples and validate this approach as a versatile means to enrich for genomes of interest in metagenomic samples. IMPORTANCE Pathogens that contaminate the food supply or spread through other means can cause outbreaks that bring devastating repercussions to the health of a populace. Investigations to trace the source of these outbreaks are initiated rapidly but can be drawn out due to the labored methods of pathogen isolation. Metagenomic sequencing can alleviate this hurdle but is often insufficiently sensitive. The approach and implementations detailed here provide a rapid means to enrich for many pathogens involved in foodborne outbreaks, thereby improving the utility of metagenomic sequencing as a tool in outbreak investigations. Additionally, this approach provides a means to broadly enrich for otherwise minute levels of modified DNA, which may escape unnoticed in metagenomic samples.
Keywords: DNA; foodborne outbreaks; metagenomics; methylation; modifications; restriction endonuclease; sequencing.
Conflict of interest statement
The authors declare a conflict of interest. A provisional patent has been filed on this technique by the authors of this manuscript.
Figures


References
-
- Buytaers FE, Saltykova A, Denayer S, Verhaegen B, Vanneste K, Roosens NHC, Piérard D, Marchal K, De Keersmaecker SCJ. 2020. A practical method to implement strain-level metagenomics-based foodborne outbreak investigation and source tracking in routine. Microorganisms 8:1191. 10.3390/microorganisms8081191. - DOI - PMC - PubMed
-
- Buytaers FE, Saltykova A, Mattheus W, Verhaegen B, Roosens NHC, Vanneste K, Laisnez V, Hammami N, Pochet B, Cantaert V, Marchal K, Denayer S, De Keersmaecker SCJ. 2021. Application of a strain-level shotgun metagenomics approach on food samples: resolution of the source of a Salmonella food-borne outbreak. Microb Genom 7:e000547. 10.1099/mgen.0.000547. - DOI - PMC - PubMed
-
- Saltykova A, Buytaers FE, Denayer S, Verhaegen B, Piérard D, Roosens NHC, Marchal K, De Keersmaecker SCJ. 2020. Strain-level metagenomic data analysis of enriched in vitro and in silico spiked food samples: paving the way towards a culture-free foodborne outbreak investigation using STEC as a case study. Int J Mol Sci 21:5688. 10.3390/ijms21165688. - DOI - PMC - PubMed
-
- Buytaers FE, Saltykova A, Denayer S, Verhaegen B, Vanneste K, Roosens NHC, Piérard D, Marchal K, De Keersmaecker SCJ. 2021. Towards real-time and affordable strain-level metagenomics-based foodborne outbreak investigations using oxford nanopore sequencing technologies. Front Microbiol 12:738284. 10.3389/fmicb.2021.738284. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
