

IL-3-zetakine combined with a CD33 costimulatory receptor as a dual CAR approach for safer and selective targeting of AML
- PMID: 36521101
- PMCID: PMC10300303
- DOI: 10.1182/bloodadvances.2022008762
IL-3-zetakine combined with a CD33 costimulatory receptor as a dual CAR approach for safer and selective targeting of AML
Abstract
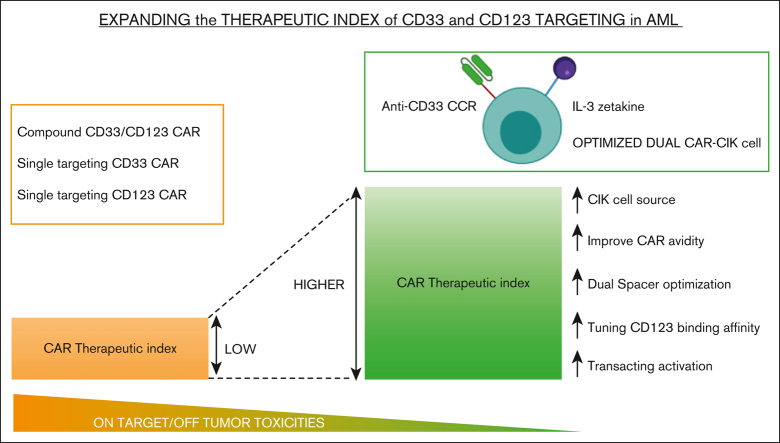
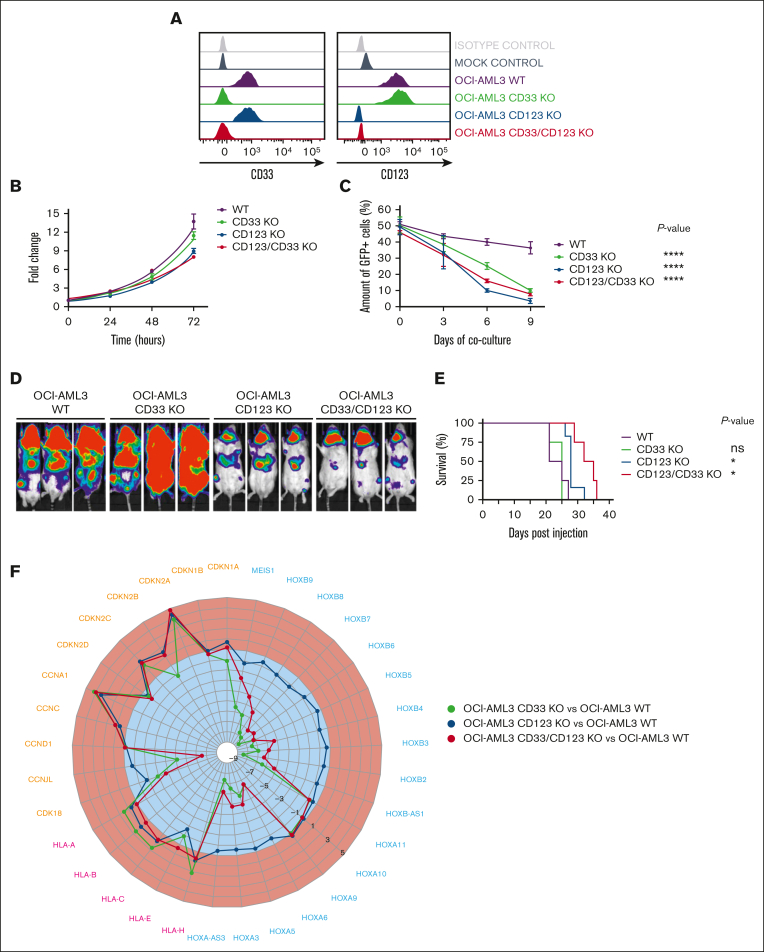
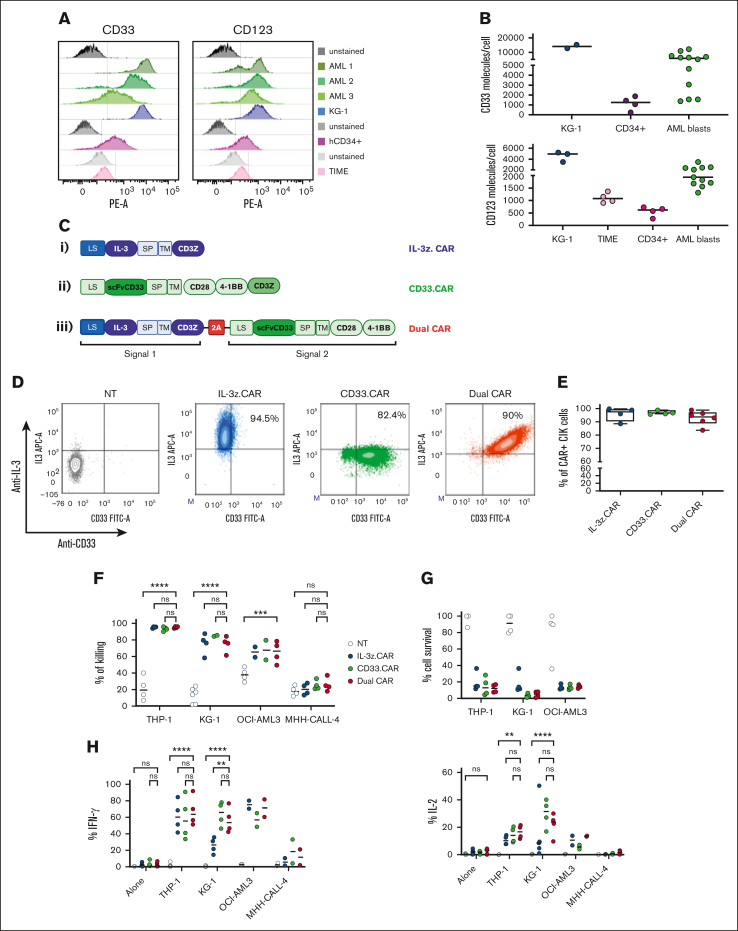
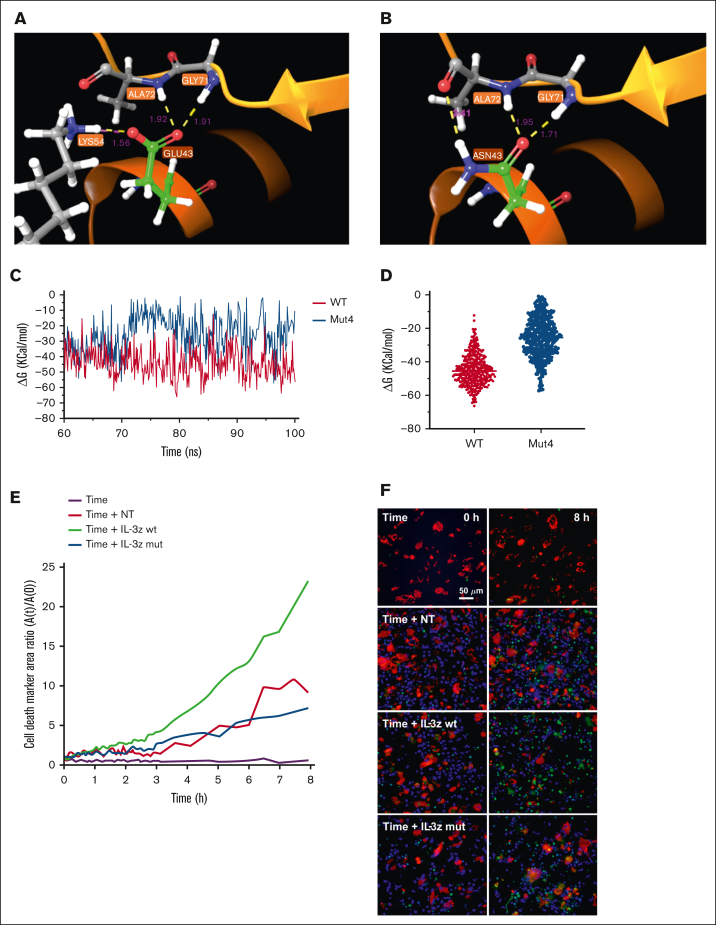
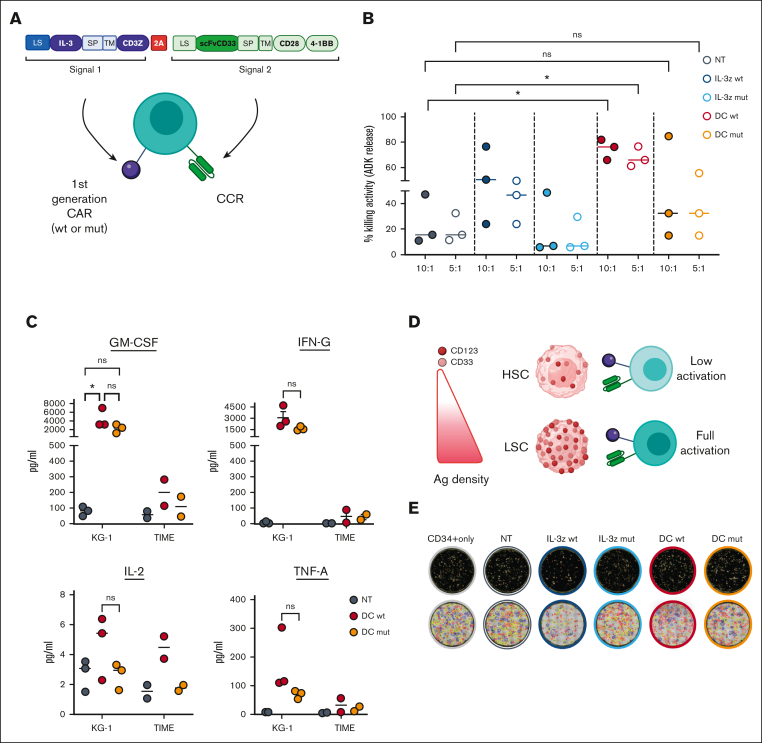
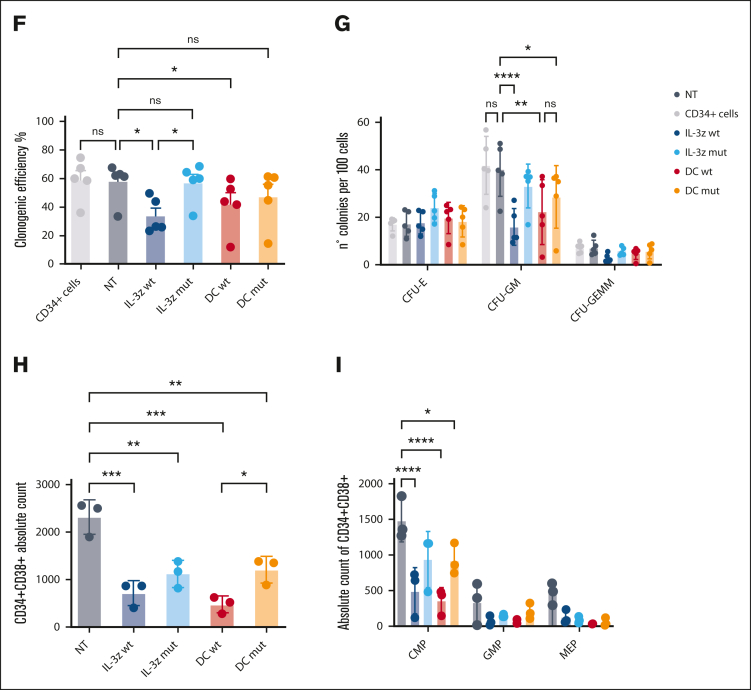
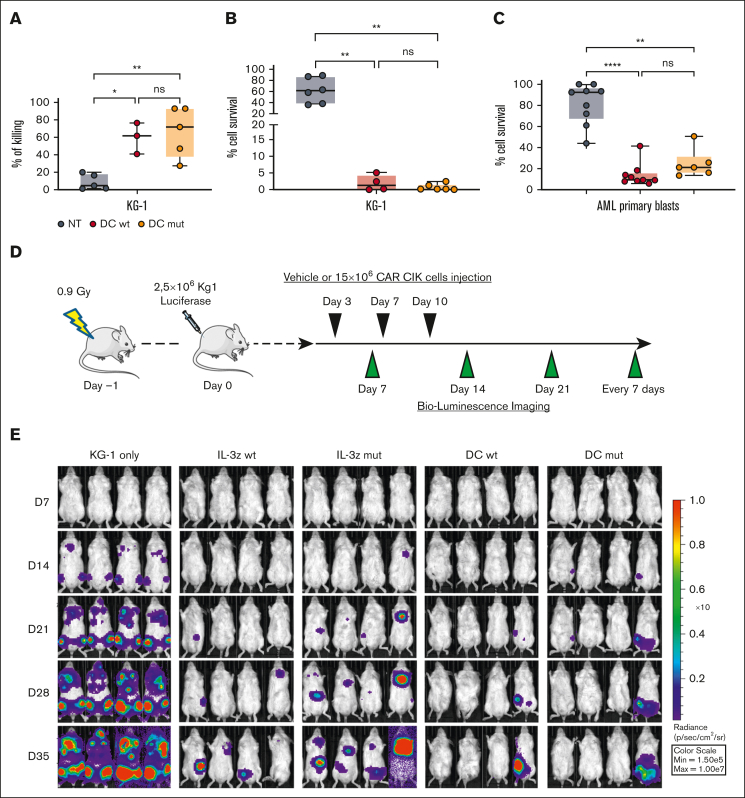
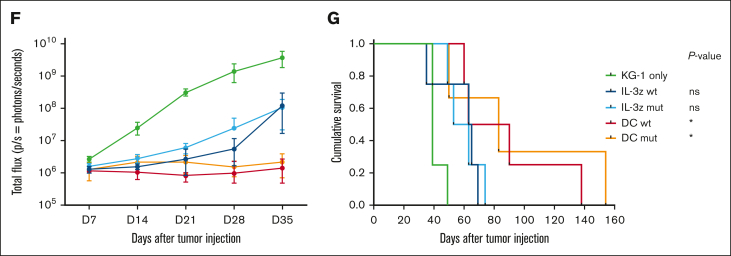
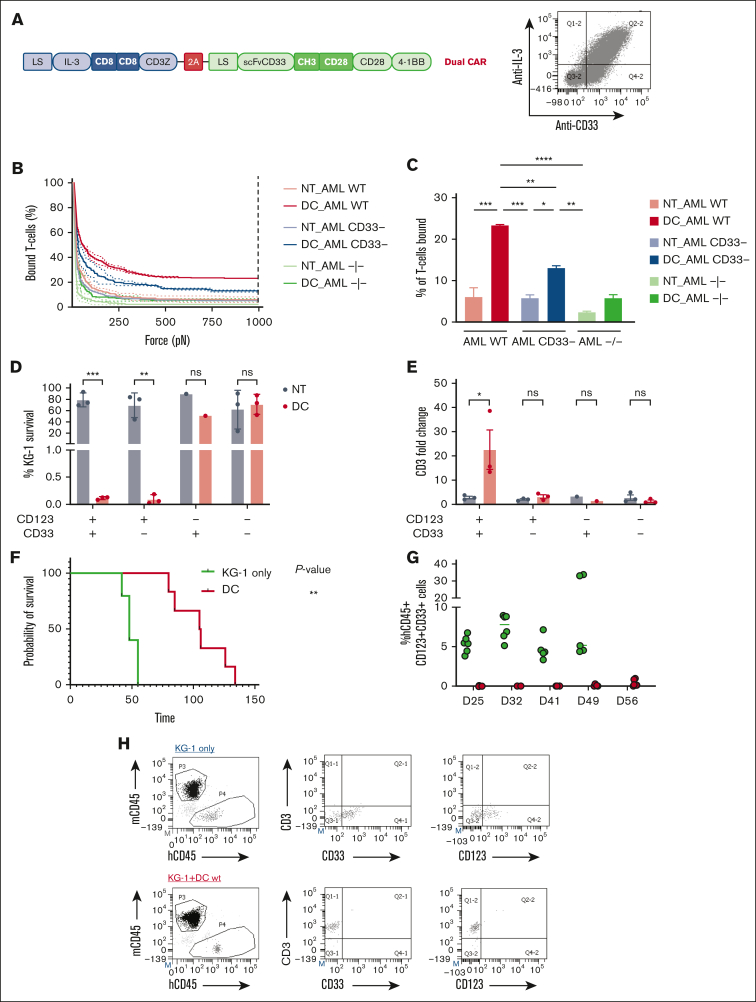
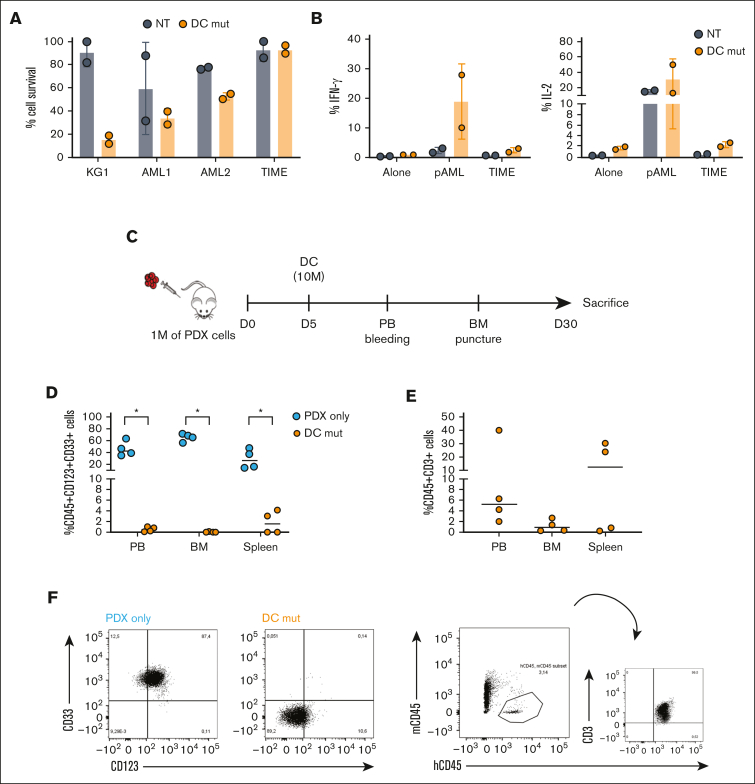
Acute myeloid leukemia (AML) still represents an unmet clinical need for adult and pediatric patients. Adoptive cell therapy by chimeric antigen receptor (CAR)-engineered T cells demonstrated a high therapeutic potential, but further development is required to ensure a safe and durable disease remission in AML, especially in elderly patients. To date, translation of CAR T-cell therapy in AML is limited by the absence of an ideal tumor-specific antigen. CD123 and CD33 are the 2 most widely overexpressed leukemic stem cell biomarkers but their shared expression with endothelial and hematopoietic stem and progenitor cells increases the risk of undesired vascular and hematologic toxicities. To counteract this issue, we established a balanced dual-CAR strategy aimed at reducing off-target toxicities while retaining full functionality against AML. Cytokine-induced killer (CIK) cells, coexpressing a first-generation low affinity anti-CD123 interleukin-3-zetakine (IL-3z) and an anti-CD33 as costimulatory receptor without activation signaling domains (CD33.CCR), demonstrated a powerful antitumor efficacy against AML targets without any relevant toxicity on hematopoietic stem and progenitor cells and endothelial cells. The proposed optimized dual-CAR cytokine-induced killer cell strategy could offer the opportunity to unleash the potential of specifically targeting CD123+/CD33+ leukemic cells while minimizing toxicity against healthy cells.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: Fondazione Tettamanti submitted a PCT application on 6 November 2015 (PCT/EP2015/075980), “Improved method for the generation of genetically modified cells,” Biondi, A., Biagi, E., Magnani, C.F., Tettamanti, S. The technology was licensed to CoImmune, Inc. for further development. M.P.M. reports honoraria from Rasna Therapeutics, Inc. for scientific advisor activities and serves as consultant for scientific advisory boards of AbbVie, Amgen, Celgene, Janssen, Novartis, Pfizer, and Jazz Pharmaceuticals. B.F. licensed a patent on NPM1 mutants (#102004901256449) and declares honoraria from Rasna Therapeutics, Inc. for scientific advisor activities. The remaining authors declare no competing financial interests.
Figures
References
-
- Heuser M, Mina A, Stein EM, Altman JK. How precision medicine is changing acute myeloid leukemia therapy. Am Soc Clin Oncol Educ Book. 2019;39:411–420. - PubMed
-
- Estey EH. Acute myeloid leukemia: 2021 update on risk-stratification and management. Am J Hematol. 2020;95(11):1368–1398. - PubMed
-
- Rambaldi A, Biagi E, Bonini C, Biondi A, Introna M. Cell-based strategies to manage leukemia relapse: efficacy and feasibility of immunotherapy approaches. Leukemia. 2015;29(1):1–10. - PubMed
-
- Martínez-Cuadrón D, Serrano J, Gil C, et al. Evolving treatment patterns and outcomes in older patients (≥60 years) with AML: changing everything to change nothing? Leukemia. 2021;35(6):1571–1585. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
