

Multi-omics identify falling LRRC15 as a COVID-19 severity marker and persistent pro-thrombotic signals in convalescence
- PMID: 36522333
- PMCID: PMC9753891
- DOI: 10.1038/s41467-022-35454-4
Multi-omics identify falling LRRC15 as a COVID-19 severity marker and persistent pro-thrombotic signals in convalescence
Abstract
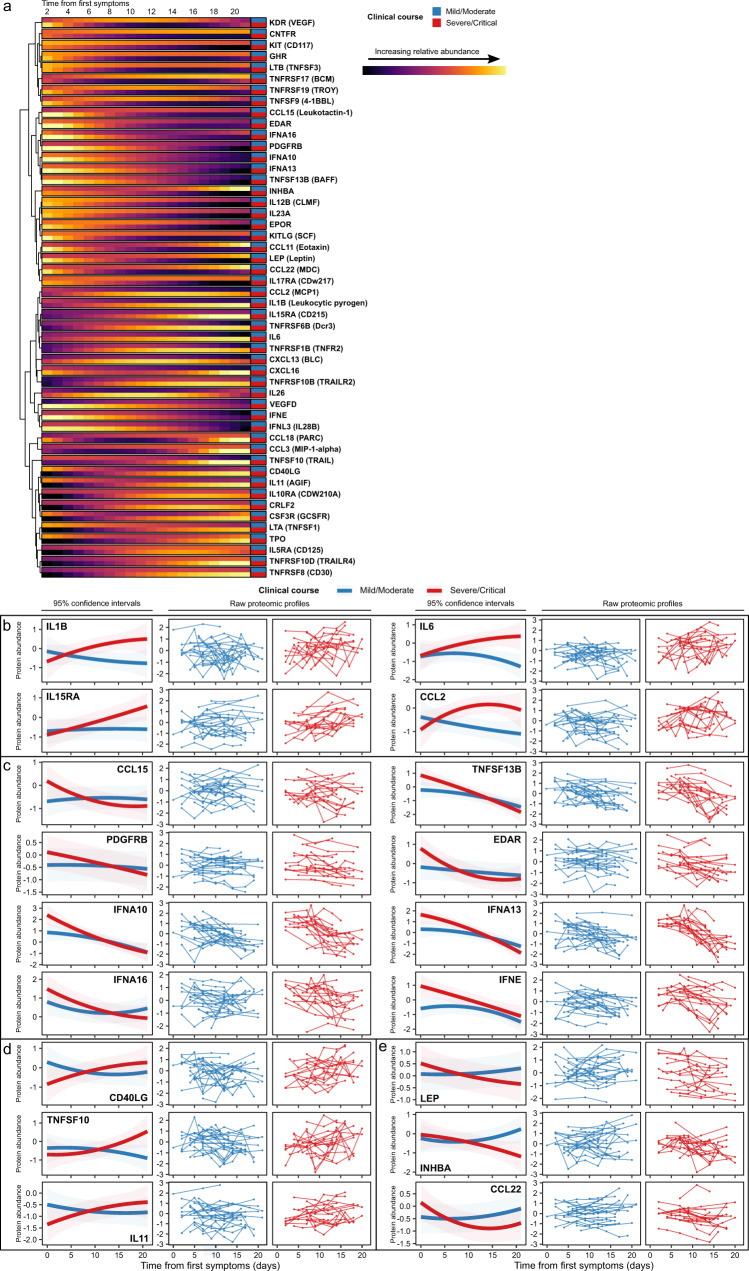
Patients with end-stage kidney disease (ESKD) are at high risk of severe COVID-19. Here, we perform longitudinal blood sampling of ESKD haemodialysis patients with COVID-19, collecting samples pre-infection, serially during infection, and after clinical recovery. Using plasma proteomics, and RNA-sequencing and flow cytometry of immune cells, we identify transcriptomic and proteomic signatures of COVID-19 severity, and find distinct temporal molecular profiles in patients with severe disease. Supervised learning reveals that the plasma proteome is a superior indicator of clinical severity than the PBMC transcriptome. We show that a decreasing trajectory of plasma LRRC15, a proposed co-receptor for SARS-CoV-2, is associated with a more severe clinical course. We observe that two months after the acute infection, patients still display dysregulated gene expression related to vascular, platelet and coagulation pathways, including PF4 (platelet factor 4), which may explain the prolonged thrombotic risk following COVID-19.
© 2022. The Author(s).
Conflict of interest statement
None of the authors have any patents (planned, pending or issued) or competing interests relevant to this work. Other interests unrelated to this work: S.P.M. reports personal fees from Celltrion, Rigel, GSK and Cello; M.C.P. reports consulting honoraria with Alexion, Apellis, Achillion, Novartis and Gyroscope; D.C.T. reports speaker and consultancy fees from Astra-Zeneca and Novartis; J.E.P. has received travel and accommodation expenses and hospitality from Olink proteomics to speak at Olink-sponsored academic meetings (none within the past 5 years). None of the other authors have any interests to declare.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
