

KMT2D deficiency drives lung squamous cell carcinoma and hypersensitivity to RTK-RAS inhibition
- PMID: 36525973
- PMCID: PMC10388706
- DOI: 10.1016/j.ccell.2022.11.015
KMT2D deficiency drives lung squamous cell carcinoma and hypersensitivity to RTK-RAS inhibition
Abstract
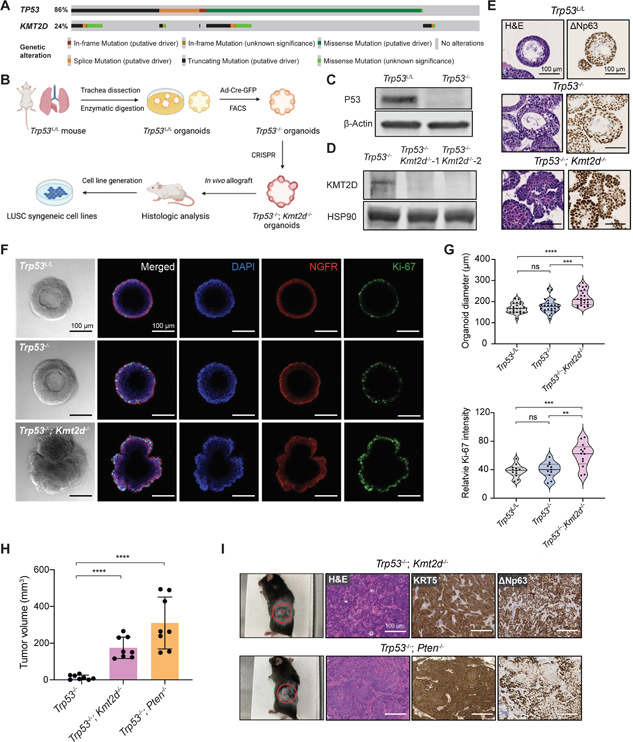
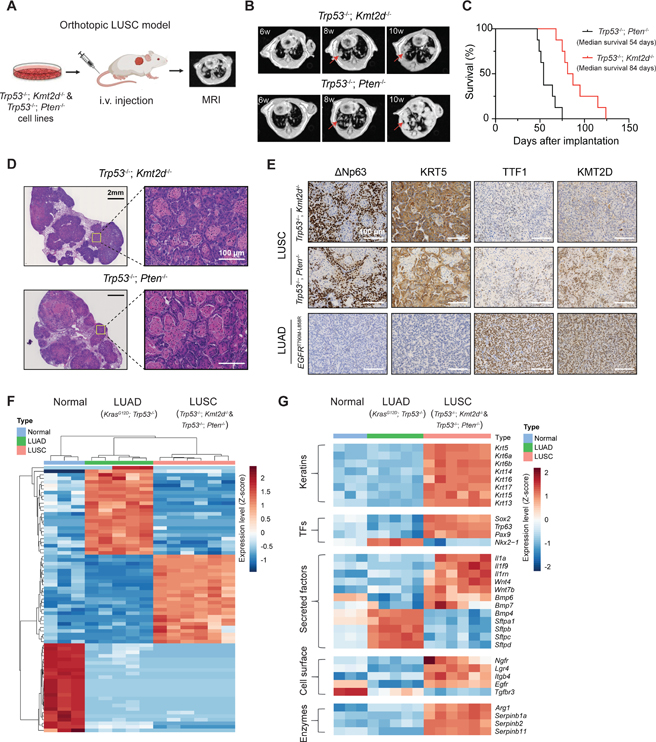
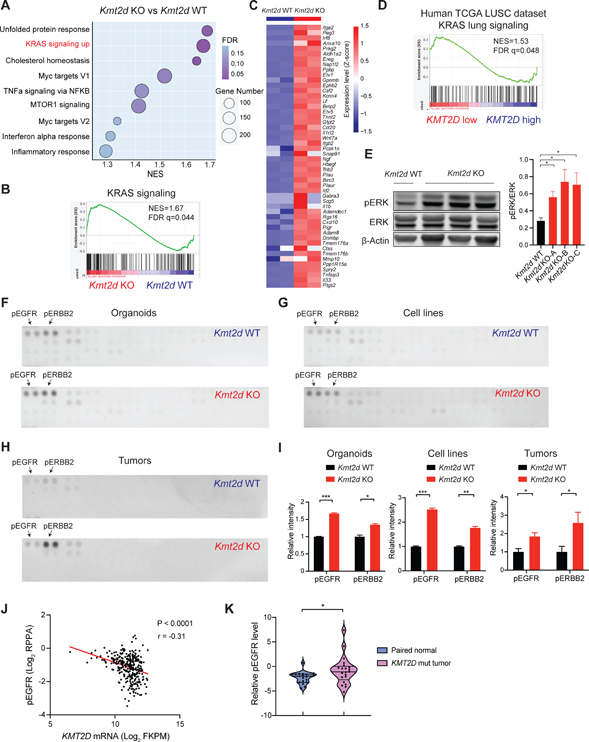
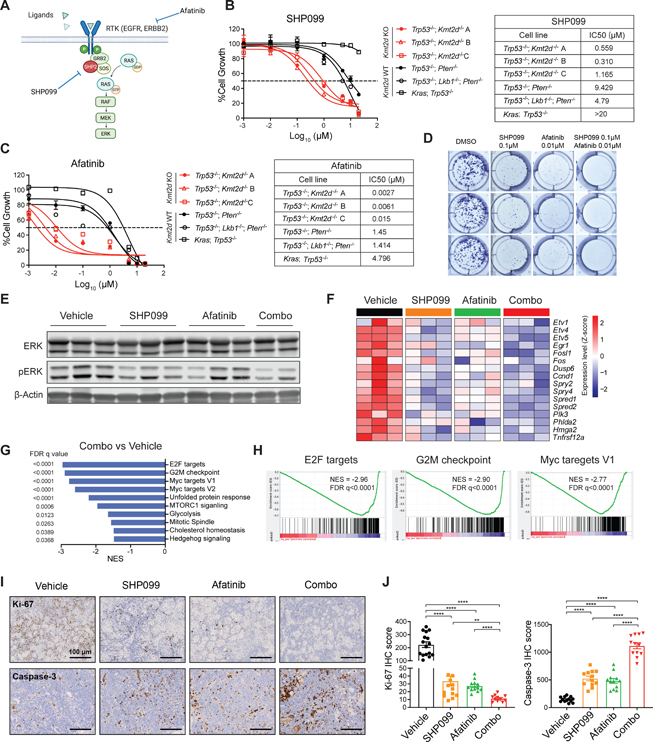
Lung squamous cell carcinoma (LUSC) represents a major subtype of lung cancer with limited treatment options. KMT2D is one of the most frequently mutated genes in LUSC (>20%), and yet its role in LUSC oncogenesis remains unknown. Here, we identify KMT2D as a key regulator of LUSC tumorigenesis wherein Kmt2d deletion transforms lung basal cell organoids to LUSC. Kmt2d loss increases activation of receptor tyrosine kinases (RTKs), EGFR and ERBB2, partly through reprogramming the chromatin landscape to repress the expression of protein tyrosine phosphatases. These events provoke a robust elevation in the oncogenic RTK-RAS signaling. Combining SHP2 inhibitor SHP099 and pan-ERBB inhibitor afatinib inhibits lung tumor growth in Kmt2d-deficient LUSC murine models and in patient-derived xenografts (PDXs) harboring KMT2D mutations. Our study identifies KMT2D as a pivotal epigenetic modulator for LUSC oncogenesis and suggests that KMT2D loss renders LUSC therapeutically vulnerable to RTK-RAS inhibition.
Keywords: EGFR; ERBB2; KMT2D; SHP2; lung squamous cell carcinoma; organoids.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.-K.W. is a founder and equity holder of G1 Therapeutics and has sponsored research agreements with Takeda, TargImmune, Bristol-Myers Squibb (BMS), Mirati, Merus, and Alkermes and consulting and sponsored research agreements with AstraZeneca, Janssen, Pfizer, Novartis, Merck, Zentalis, BridgeBio, and Blueprint. A.J.B. has received funding from Bayer, Novartis, Merck, and Repare and is a co-founder with equity in Signet Therapeutics. Y.P., H.Han., H.Z., and K.-K.W. have ownership interest in a patent application.
Figures
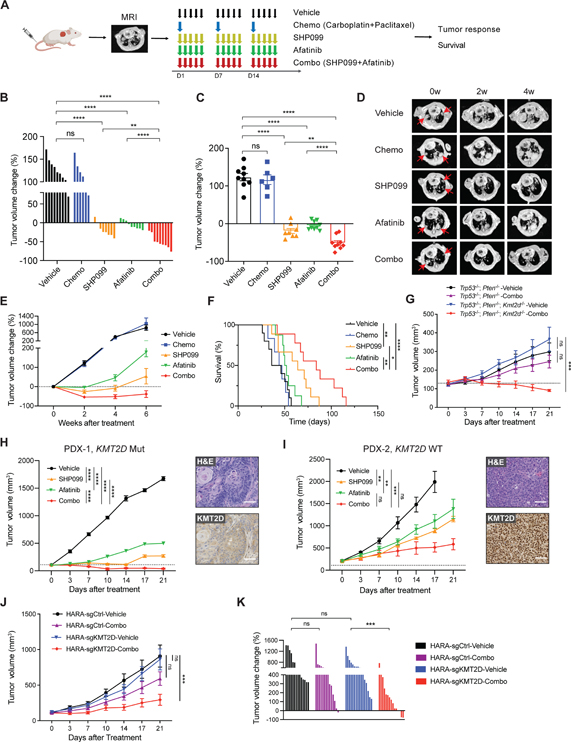
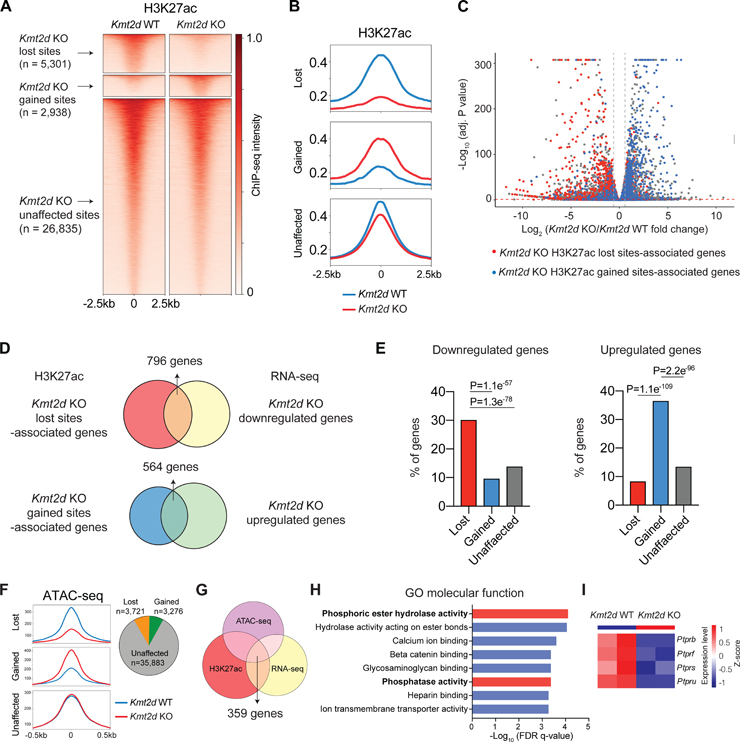
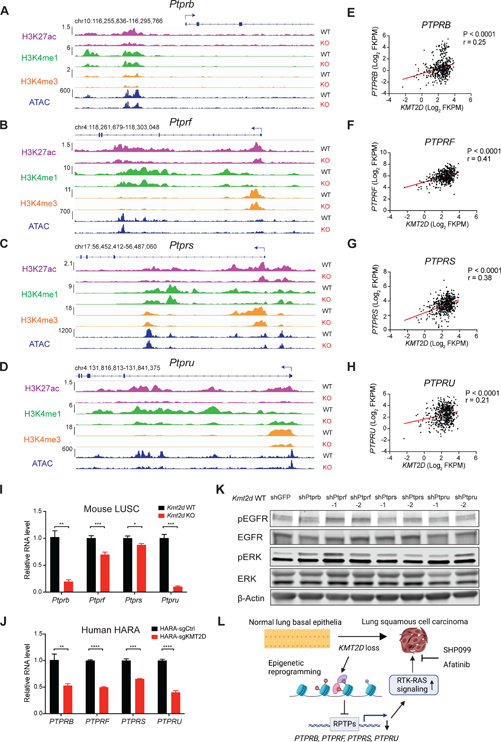
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
