

Autosomal Dominant Tubulointerstitial Kidney Disease: An Emerging Cause of Genetic CKD
- PMID: 36531871
- PMCID: PMC9751576
- DOI: 10.1016/j.ekir.2022.08.012
Autosomal Dominant Tubulointerstitial Kidney Disease: An Emerging Cause of Genetic CKD
Abstract
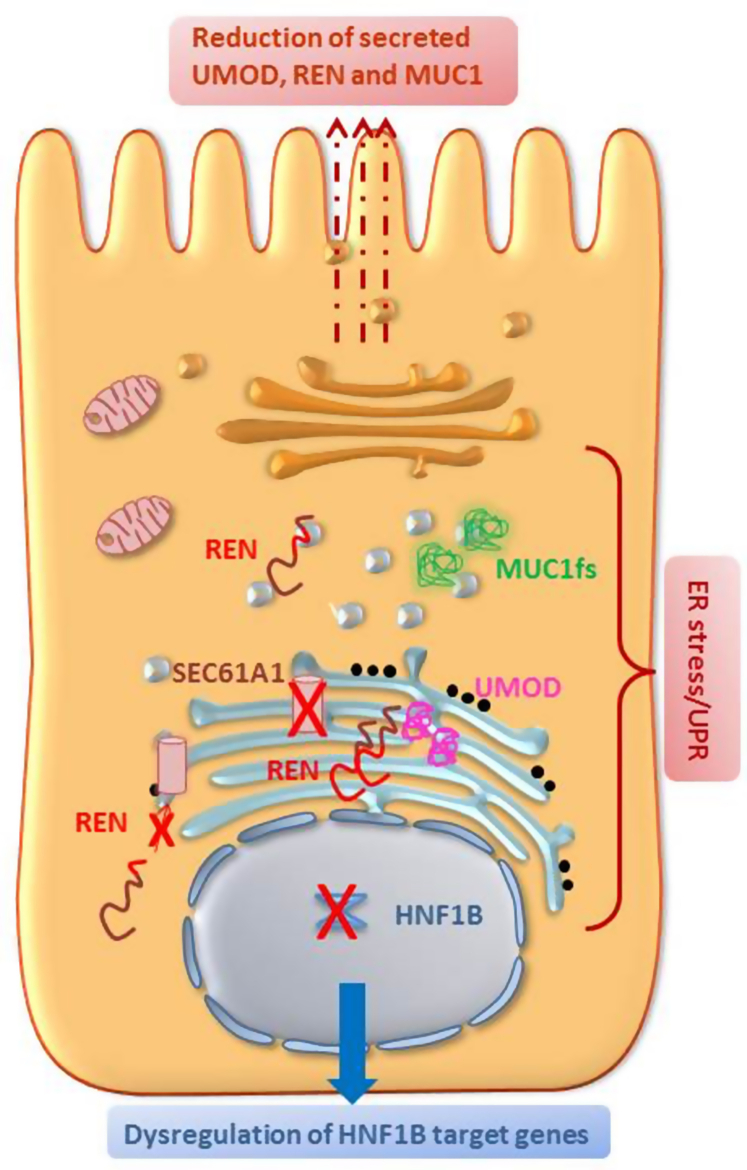
Autosomal dominant tubulointerstitial kidney disease (ADTKD) is a rare inherited disorder characterized by progressive loss of kidney function, nonsignificant urinalysis and tubulointerstitial fibrosis. ADTKD progresses to end stage renal disease (ESRD) in adulthood. The classification of ADTKD is an evolving concept and the agreement is now that, due to the overlap in terms of phenotype characteristics, this should be based on the involved gene. The umbrella term ADTKD therefore includes different conditions as follows: ADTKD-UMOD, ADKTD-MUC1, ADTKD-REN, and ADTK-HNF1B, with ADTKD-SEC61A1 and ADTKD-DNAJB11 as a further rare and atypical diagnosis recently described. The employment of next-generation sequencing (NGS) as a diagnostic tool in patients with familial kidney disease has improved the diagnostic accuracy in this field with ADTKD now being considered the third genetic cause of renal disease worldwide after autosomal dominant polycystic kidney disease (ADPKD) and Alport syndrome. On average, the disease pathogenesis is similar across the different subtypes, With the exception of HNF1B, the different mutated genes give rise to misfolded proteins leading to cellular stress and cytotoxicity. Research is now focused in better defining the underlying mechanism of fibrosis to guide therapeutic interventions. The aim of this review is to discuss how the knowledge of ADTKD has evolved in the last decades, with emphasis on the clinical features, molecular diagnosis, and pathogenic aspects of the different diseases included under the ADTKD term.
Keywords: ADTKD; DNAJB11; HNF1B; MUC1; REN; UMOD.
© 2022 Published by Elsevier, Inc., on behalf of the International Society of Nephrology.
Figures



References
-
- Smith C.H., Graham J.B. Congenital medullary cysts of the kidneys with severe refractory anemia. Am J Dis Child. 1945;69:369–377.
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
