

The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK
- PMID: 36533153
- PMCID: PMC9752794
- DOI: 10.1093/ve/veac080
The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK
Erratum in
-
Correction to: The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK.Virus Evol. 2022 Dec 30;8(2):veac119. doi: 10.1093/ve/veac119. eCollection 2022. Virus Evol. 2022. PMID: 36601301 Free PMC article.
Abstract
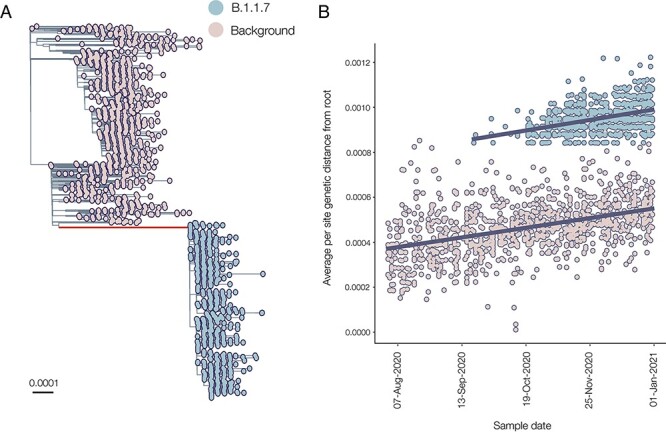
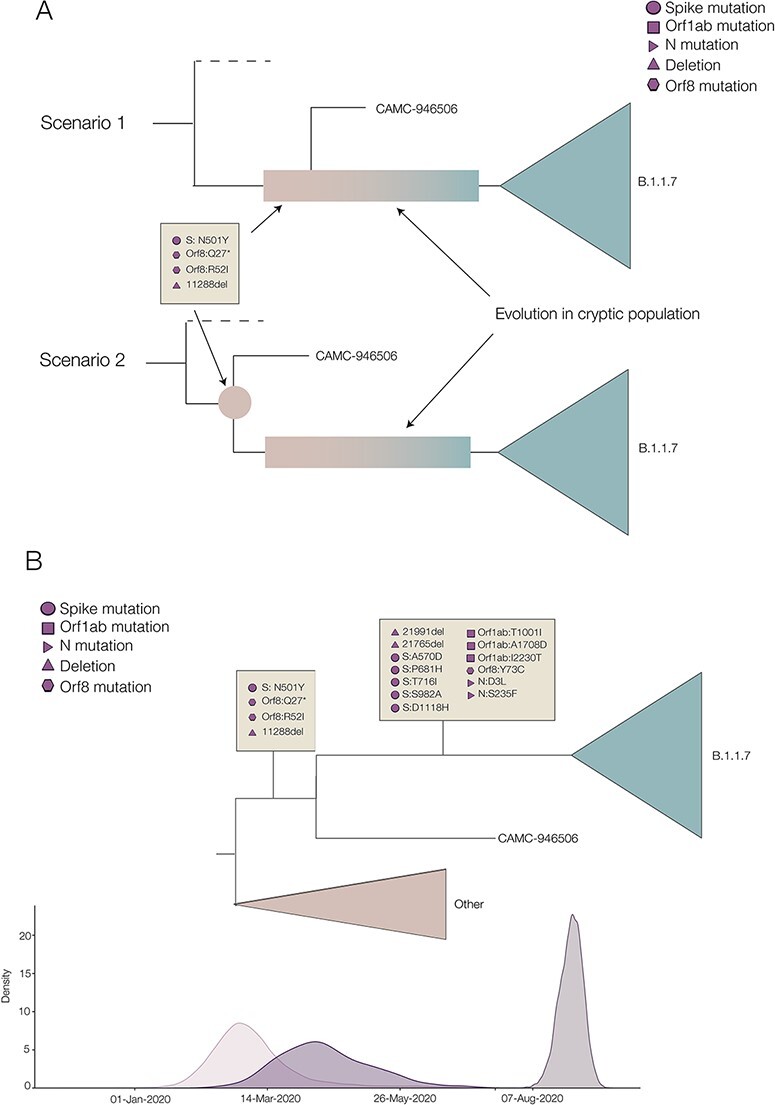
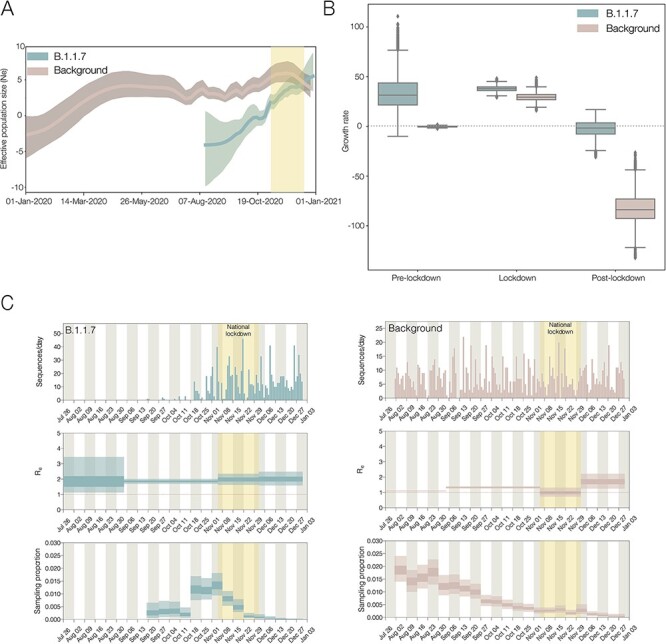
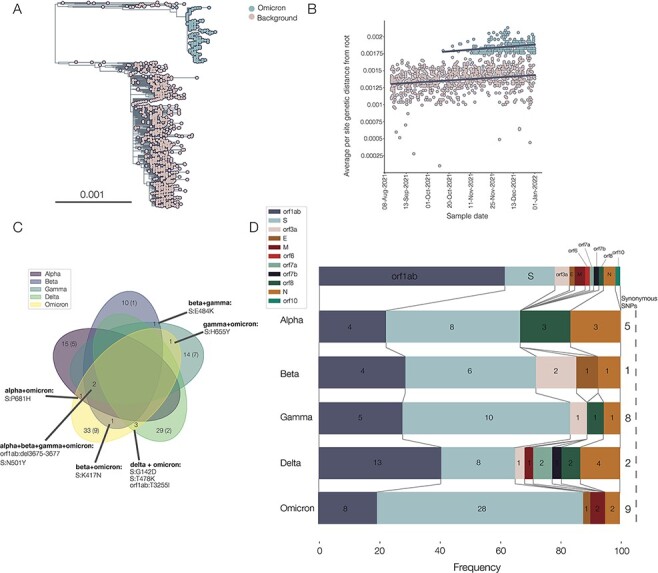
The first SARS-CoV-2 variant of concern (VOC) to be designated was lineage B.1.1.7, later labelled by the World Health Organization as Alpha. Originating in early autumn but discovered in December 2020, it spread rapidly and caused large waves of infections worldwide. The Alpha variant is notable for being defined by a long ancestral phylogenetic branch with an increased evolutionary rate, along which only two sequences have been sampled. Alpha genomes comprise a well-supported monophyletic clade within which the evolutionary rate is typical of SARS-CoV-2. The Alpha epidemic continued to grow despite the continued restrictions on social mixing across the UK and the imposition of new restrictions, in particular, the English national lockdown in November 2020. While these interventions succeeded in reducing the absolute number of cases, the impact of these non-pharmaceutical interventions was predominantly to drive the decline of the SARS-CoV-2 lineages that preceded Alpha. We investigate the only two sampled sequences that fall on the branch ancestral to Alpha. We find that one is likely to be a true intermediate sequence, providing information about the order of mutational events that led to Alpha. We explore alternate hypotheses that can explain how Alpha acquired a large number of mutations yet remained largely unobserved in a region of high genomic surveillance: an under-sampled geographical location, a non-human animal population, or a chronically infected individual. We conclude that the latter provides the best explanation of the observed behaviour and dynamics of the variant, although the individual need not be immunocompromised, as persistently infected immunocompetent hosts also display a higher within-host rate of evolution. Finally, we compare the ancestral branches and mutation profiles of other VOCs and find that Delta appears to be an outlier both in terms of the genomic locations of its defining mutations and a lack of the rapid evolutionary rate on its ancestral branch. As new variants, such as Omicron, continue to evolve (potentially through similar mechanisms), it remains important to investigate the origins of other variants to identify ways to potentially disrupt their evolution and emergence.
Keywords: SARS-COV-2; Virus evolution; Within-host evolution; variants of concern.
© The Author(s) 2022. Published by Oxford University Press.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
