

AP1/Fra1 confers resistance to MAPK cascade inhibition in pancreatic cancer
- PMID: 36534167
- PMCID: PMC9763154
- DOI: 10.1007/s00018-022-04638-y
AP1/Fra1 confers resistance to MAPK cascade inhibition in pancreatic cancer
Abstract
Targeting KRAS downstream signaling remains an important therapeutic approach in pancreatic cancer. We used primary pancreatic ductal epithelial cells and mouse models allowing the conditional expression of oncogenic KrasG12D, to investigate KRAS signaling integrators. We observed that the AP1 family member FRA1 is tightly linked to the KRAS signal and expressed in pre-malignant lesions and the basal-like subtype of pancreatic cancer. However, genetic-loss-of-function experiments revealed that FRA1 is dispensable for KrasG12D-induced pancreatic cancer development in mice. Using FRA1 gain- and loss-of-function models in an unbiased drug screen, we observed that FRA1 is a modulator of the responsiveness of pancreatic cancer to inhibitors of the RAF-MEK-ERK cascade. Mechanistically, context-dependent FRA1-associated adaptive rewiring of oncogenic ERK signaling was observed and correlated with sensitivity to inhibitors of canonical KRAS signaling. Furthermore, pharmacological-induced degradation of FRA1 synergizes with MEK inhibitors. Our studies establish FRA1 as a part of the molecular machinery controlling sensitivity to MAPK cascade inhibition allowing the development of mechanism-based therapies.
Keywords: AP1; ERK; FRA1; KRAS; MEK; Pancreatic cancer.
© 2022. The Author(s).
Conflict of interest statement
There are no conflicts of interest.
Figures
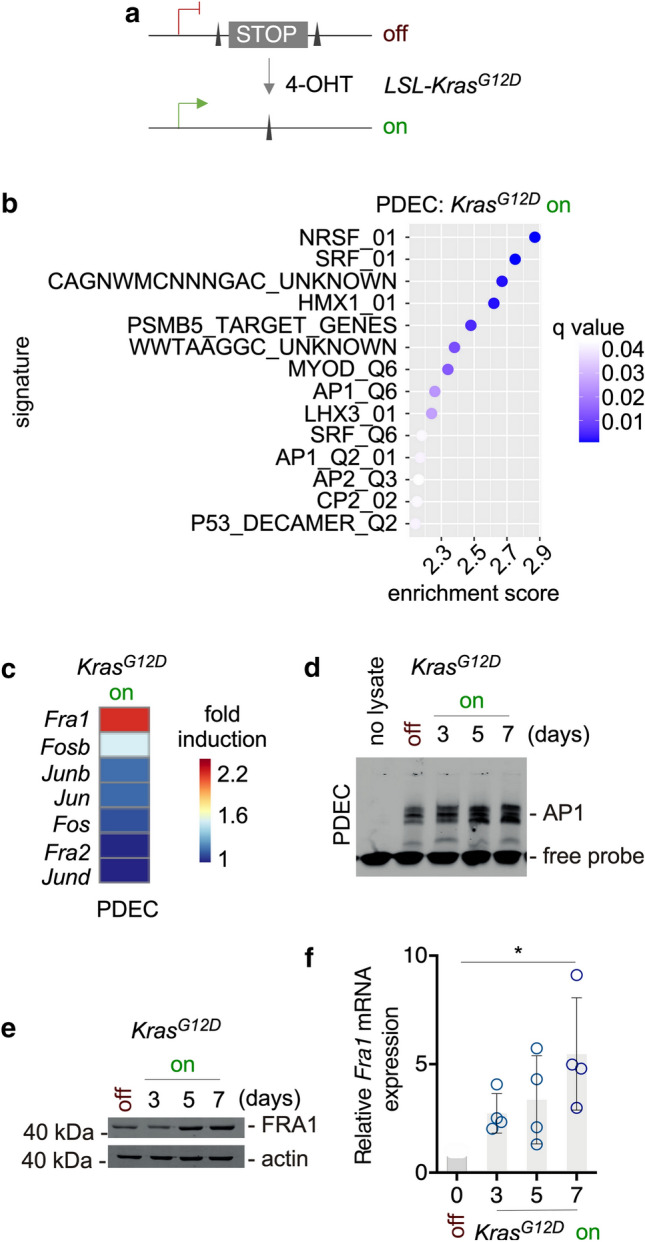
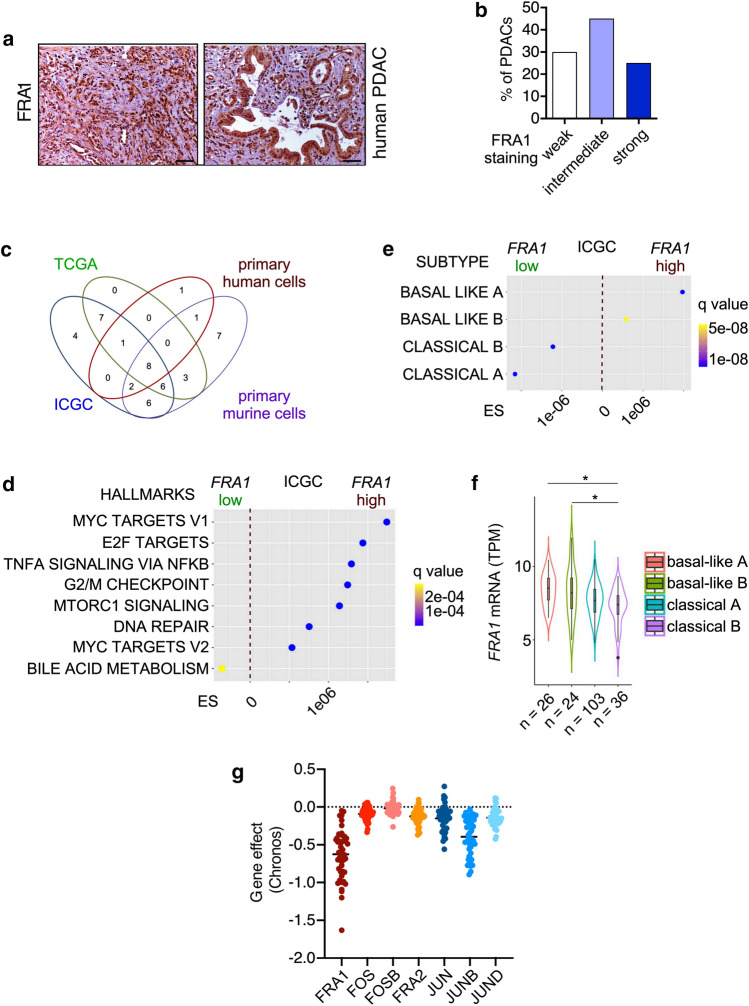
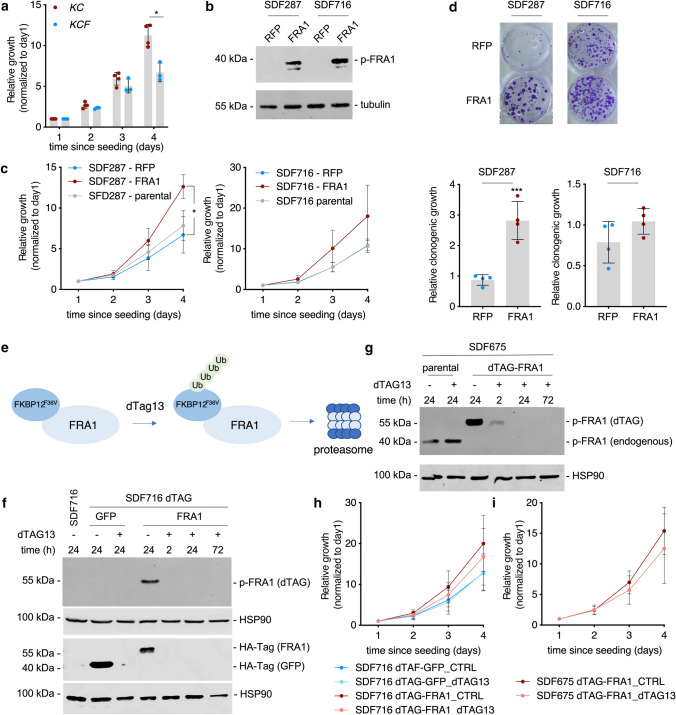
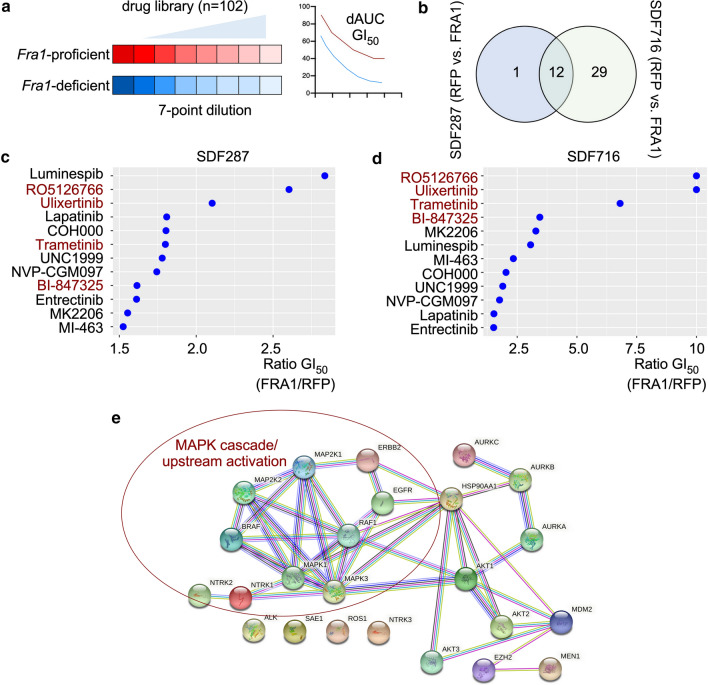

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
