

Hepatocyte Smoothened Activity Controls Susceptibility to Insulin Resistance and Nonalcoholic Fatty Liver Disease
- PMID: 36535507
- PMCID: PMC9957752
- DOI: 10.1016/j.jcmgh.2022.12.008
Hepatocyte Smoothened Activity Controls Susceptibility to Insulin Resistance and Nonalcoholic Fatty Liver Disease
Abstract
Background & aims: Nonalcoholic steatohepatitis (NASH), a leading cause of cirrhosis, strongly associates with the metabolic syndrome, an insulin-resistant proinflammatory state that disrupts energy balance and promotes progressive liver degeneration. We aimed to define the role of Smoothened (Smo), an obligatory component of the Hedgehog signaling pathway, in controlling hepatocyte metabolic homeostasis and, thereby, susceptibility to NASH.
Methods: We conditionally deleted Smo in hepatocytes of healthy chow-fed mice and performed metabolic phenotyping, coupled with single-cell RNA sequencing (RNA-seq), to characterize the role of hepatocyte Smo in regulating basal hepatic and systemic metabolic homeostasis. Liver RNA-seq datasets from 2 large human cohorts were also analyzed to define the relationship between Smo and NASH susceptibility in people.
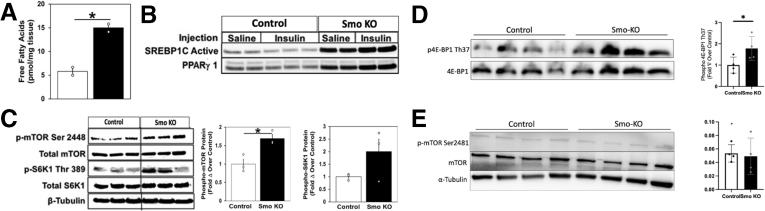
Results: Hepatocyte Smo deletion inhibited the Hedgehog pathway and promoted fatty liver, hyperinsulinemia, and insulin resistance. We identified a plausible mechanism whereby inactivation of Smo stimulated the mTORC1-SREBP1c signaling axis, which promoted lipogenesis while inhibiting the hepatic insulin cascade. Transcriptomics of bulk and single Smo-deficient hepatocytes supported suppression of insulin signaling and also revealed molecular abnormalities associated with oxidative stress and mitochondrial dysfunction. Analysis of human bulk RNA-seq data revealed that Smo expression was (1) highest in healthy livers, (2) lower in livers with NASH than in those with simple steatosis, (3) negatively correlated with markers of insulin resistance and liver injury, and (4) declined progressively as fibrosis severity worsened.
Conclusions: The Hedgehog pathway controls insulin sensitivity and energy homeostasis in adult livers. Loss of hepatocyte Hedgehog activity induces hepatic and systemic metabolic stress and enhances susceptibility to NASH by promoting hepatic lipoxicity and insulin resistance.
Keywords: hedgehog; metabolic syndrome; nonalcoholic fatty liver disease.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Figures













References
-
- Younossi Z., Anstee Q.M., Marietti M., et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15:11–20. - PubMed
-
- Eslam M., Sanyal A.J., George J., International Consensus Panel MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158:1999–2014.e1. - PubMed
-
- Ramachandran P., Matchett K.P., Dobie R., et al. Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat Rev Gastroenterol Hepatol. 2020;17:457–472. - PubMed
-
- Planas-Paz L., Orsini V., Boulter L., et al. The RSPO-LGR4/5-ZNRF3/RNF43 module controls liver zonation and size. Nat Cell Biol. 2016;18:467–479. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
