

The complement system and human autoimmune diseases
- PMID: 36535812
- PMCID: PMC10276174
- DOI: 10.1016/j.jaut.2022.102979
The complement system and human autoimmune diseases
Abstract
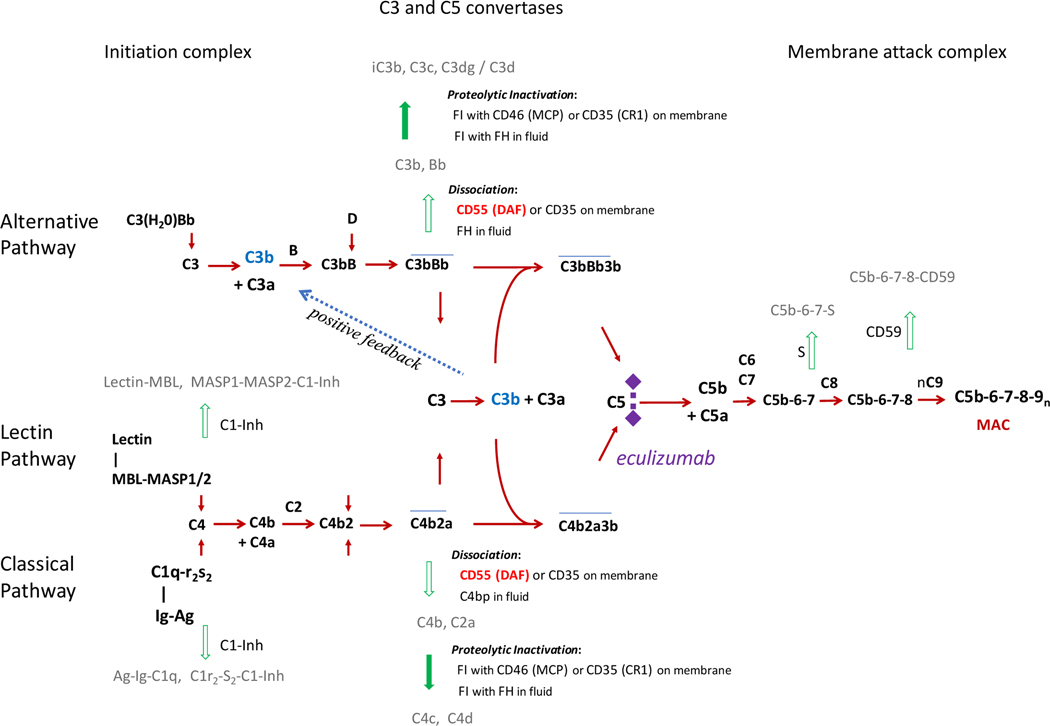
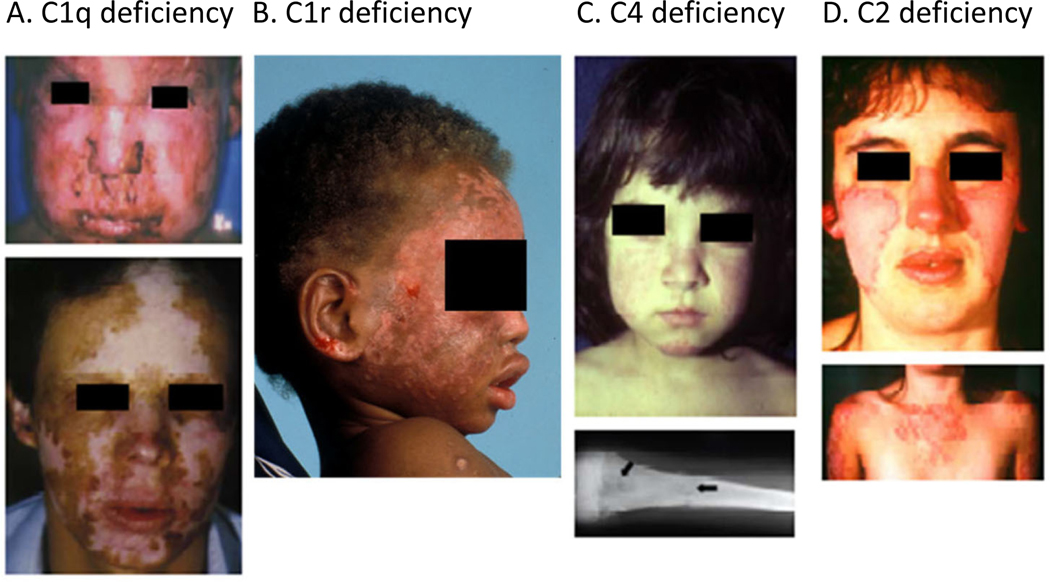
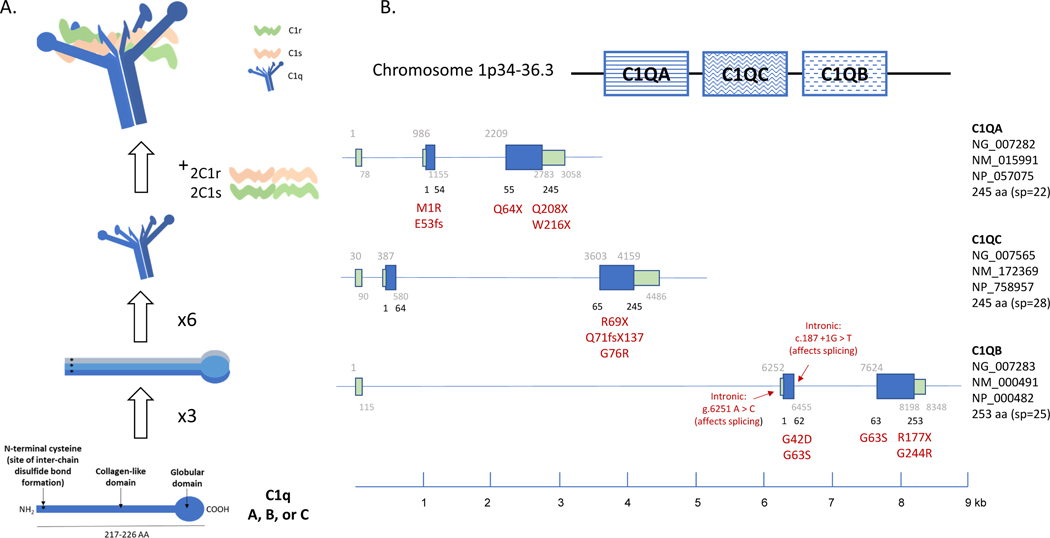
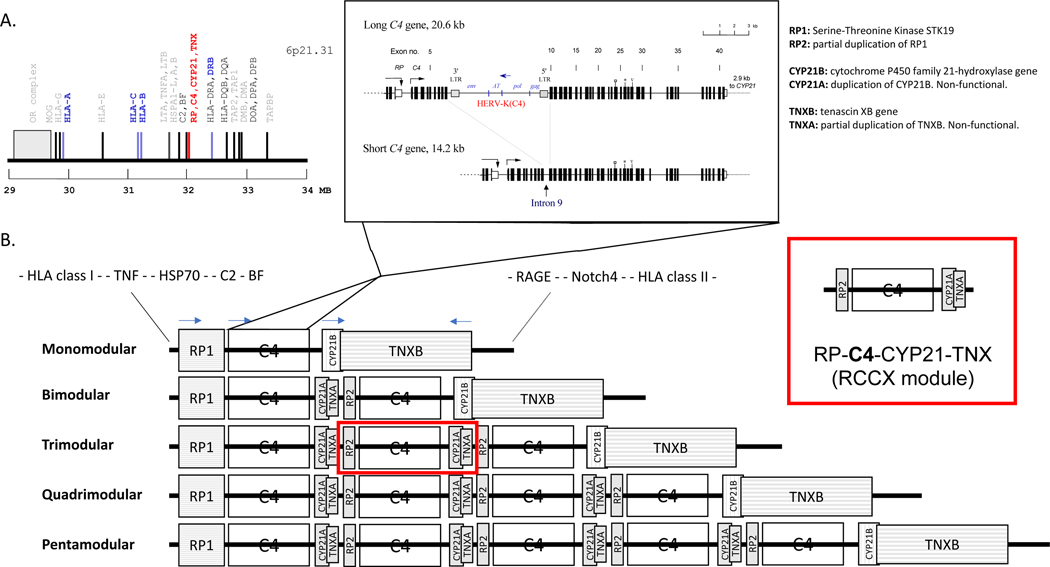
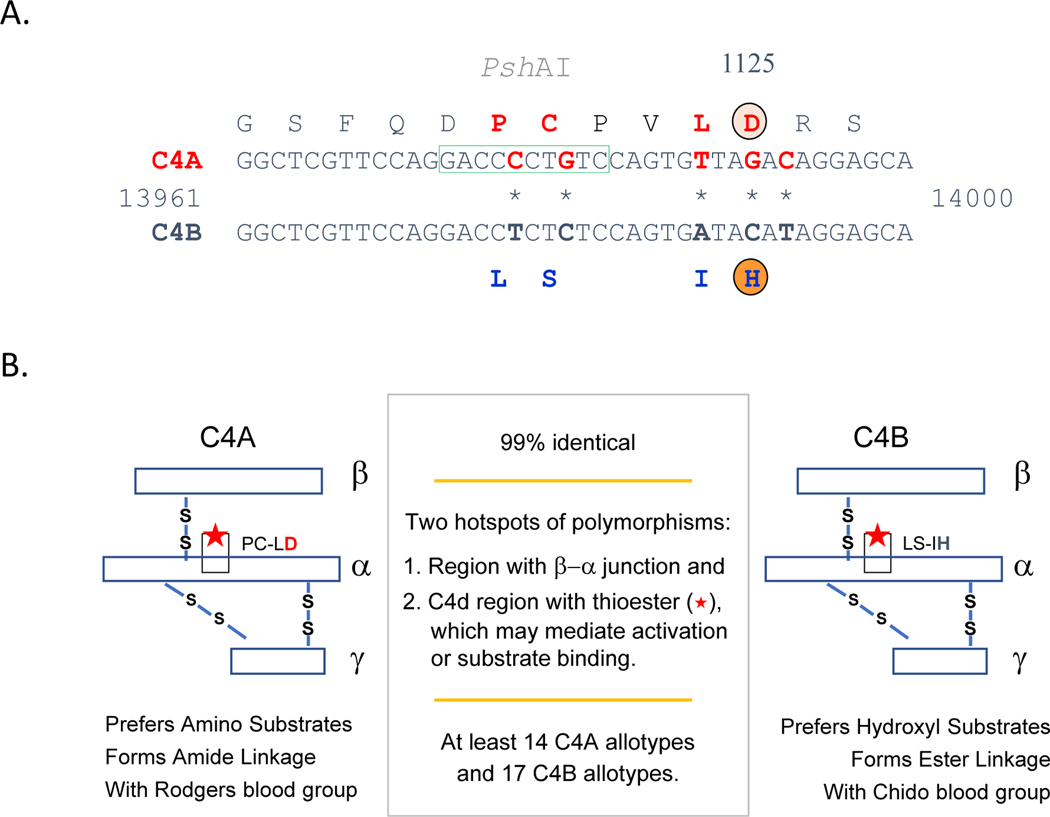
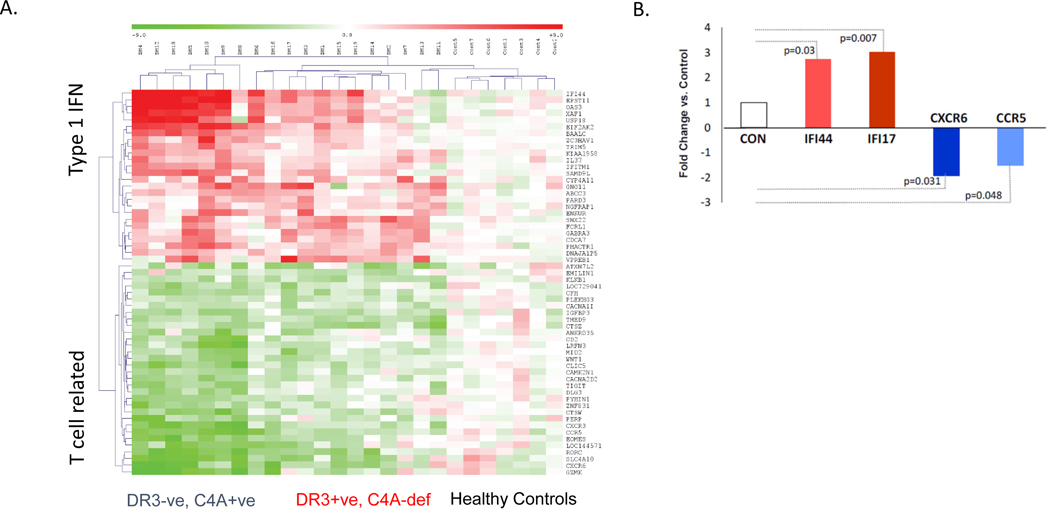
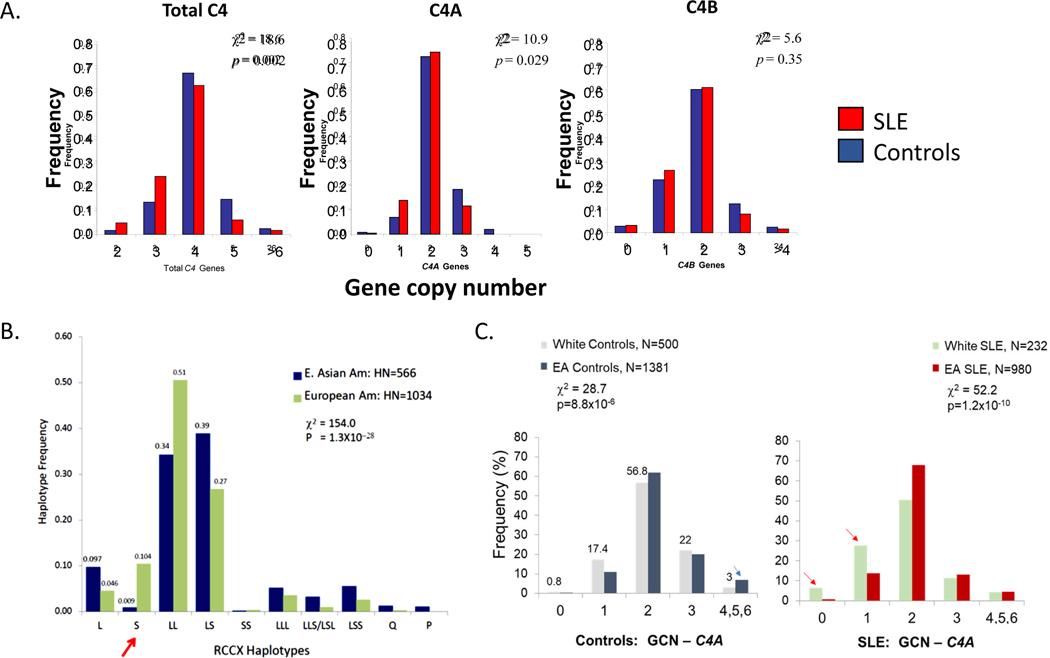
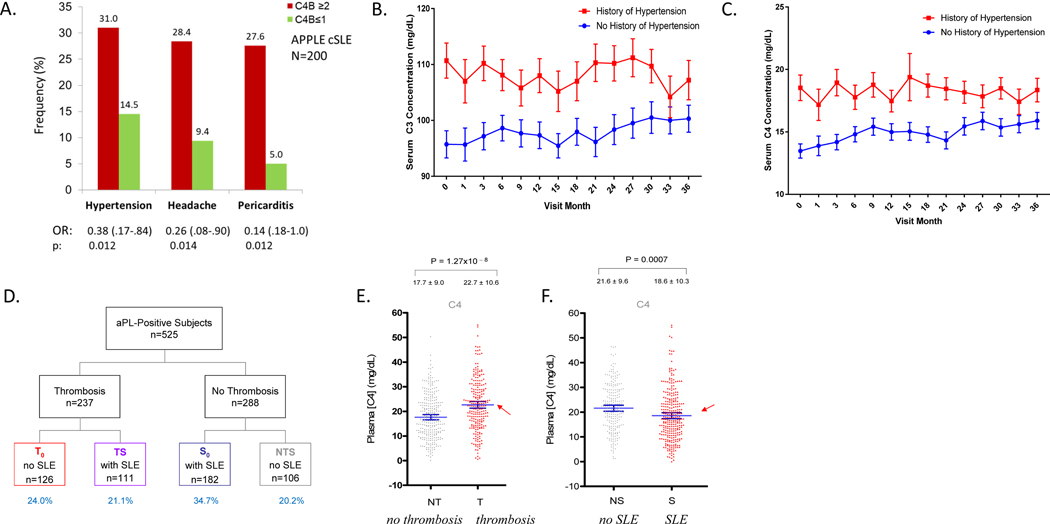
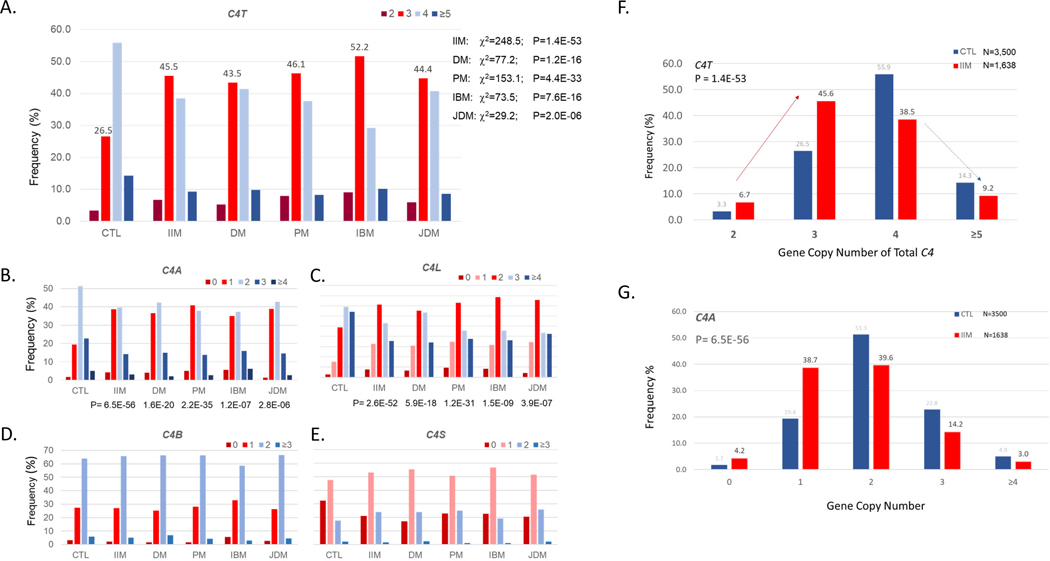
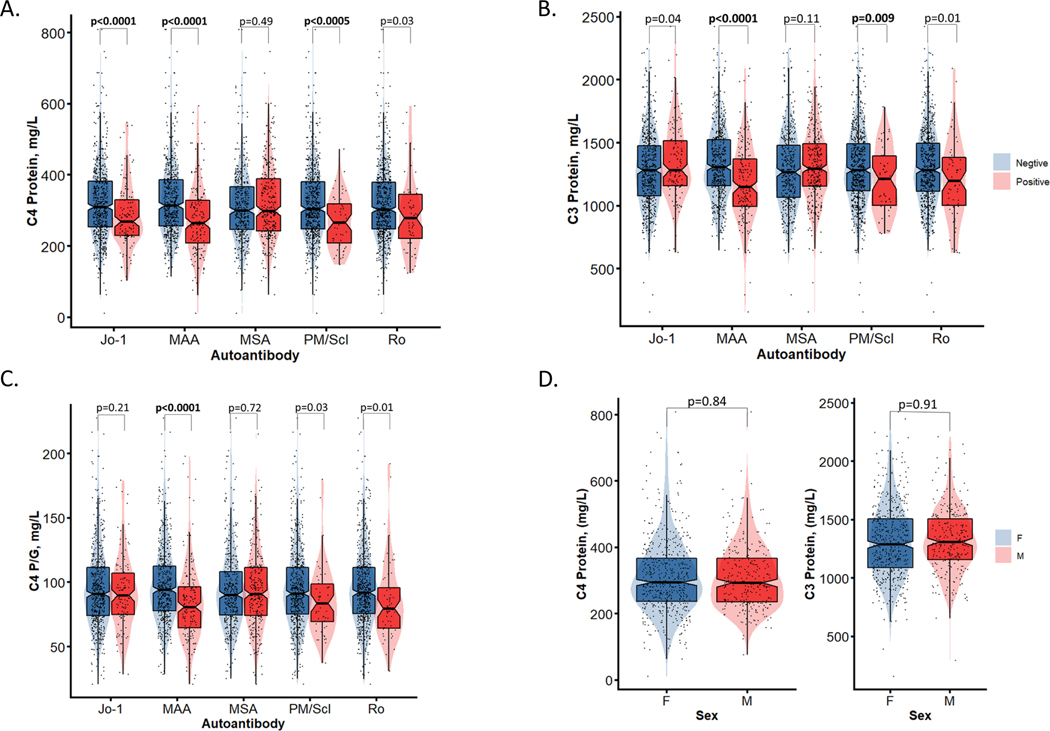
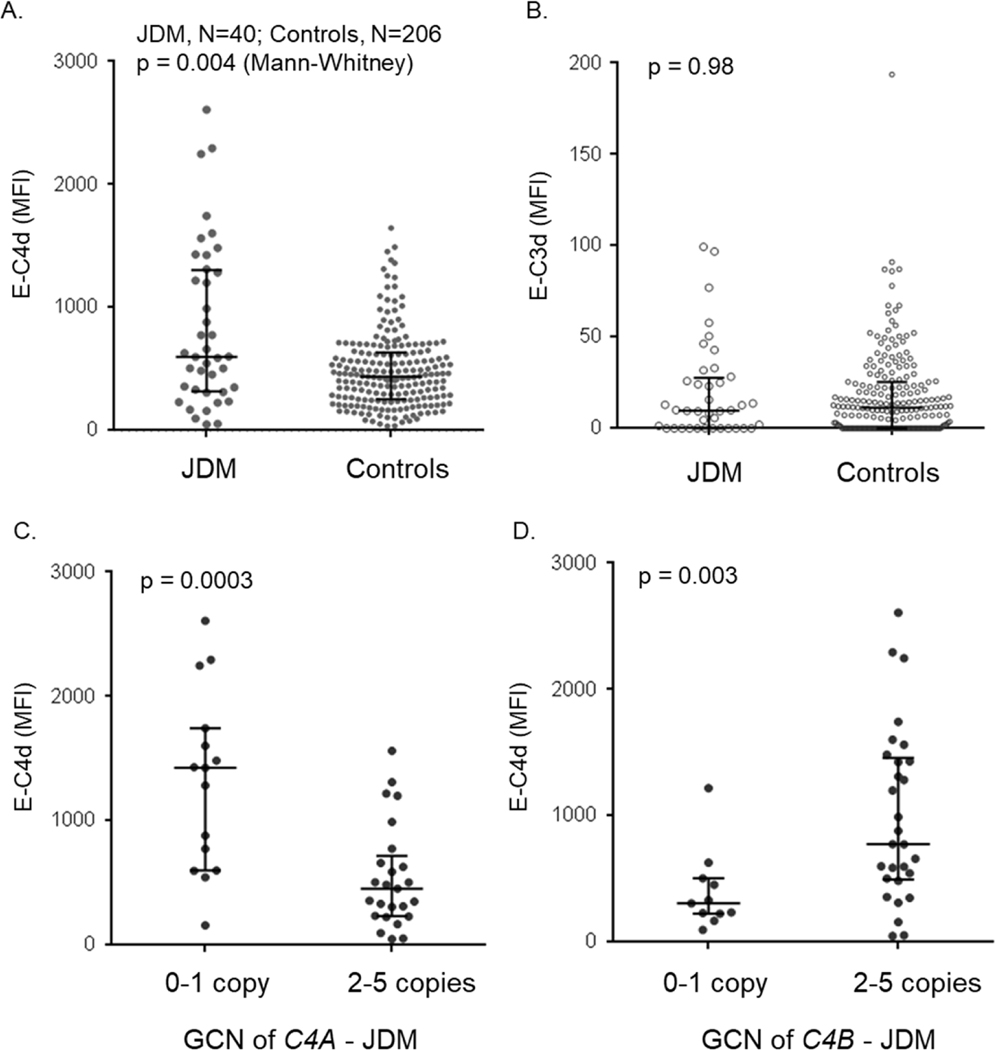
Genetic deficiencies of early components of the classical complement activation pathway (especially C1q, r, s, and C4) are the strongest monogenic causal factors for the prototypic autoimmune disease systemic lupus erythematosus (SLE), but their prevalence is extremely rare. In contrast, isotype genetic deficiency of C4A and acquired deficiency of C1q by autoantibodies are frequent among patients with SLE. Here we review the genetic basis of complement deficiencies in autoimmune disease, discuss the complex genetic diversity seen in complement C4 and its association with autoimmune disease, provide guidance as to when clinicians should suspect and test for complement deficiencies, and outline the current understanding of the mechanisms relating complement deficiencies to autoimmunity. We focus primarily on SLE, as the role of complement in SLE is well-established, but will also discuss other informative diseases such as inflammatory arthritis and myositis.
Keywords: Antiphospholipid syndrome; Autoantibodies; Classical pathway; Complement; Gene copy number variations; Genetic and acquired deficiencies; Idiopathic inflammatory myopathies; Juvenile dermatomyositis; Polymorphisms; Systemic lupus erythematosus; juvenile idiopathic arthritis; rheumatoid arthritis; type I interferon induced gene expression.
Copyright © 2022 The Authors. Published by Elsevier Ltd.. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Porter RR. Complement polymorphism, the major histocompatibility complex and associated diseases: a speculation. Mol Biol Med. 1983;1(1):161–8. Epub 1983/07/01. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
