

A comparison of the genes and genesets identified by GWAS and EWAS of fifteen complex traits
- PMID: 36535946
- PMCID: PMC9763500
- DOI: 10.1038/s41467-022-35037-3
A comparison of the genes and genesets identified by GWAS and EWAS of fifteen complex traits
Abstract
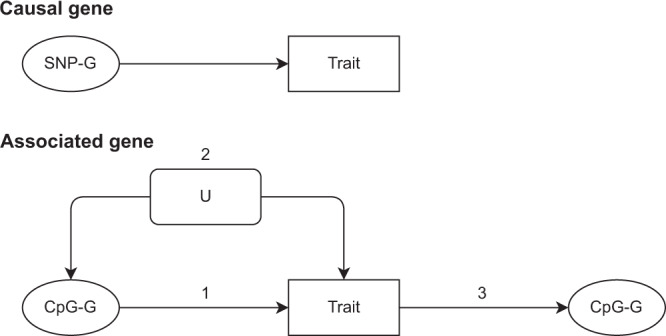
Identifying genomic regions pertinent to complex traits is a common goal of genome-wide and epigenome-wide association studies (GWAS and EWAS). GWAS identify causal genetic variants, directly or via linkage disequilibrium, and EWAS identify variation in DNA methylation associated with a trait. While GWAS in principle will only detect variants due to causal genes, EWAS can also identify genes via confounding, or reverse causation. We systematically compare GWAS (N > 50,000) and EWAS (N > 4500) results of 15 complex traits. We evaluate if the genes or gene ontology terms flagged by GWAS and EWAS overlap, and find substantial overlap for diastolic blood pressure, (gene overlap P = 5.2 × 10-6; term overlap P = 0.001). We superimpose our empirical findings against simulated models of varying genetic and epigenetic architectures and observe that in most cases GWAS and EWAS are likely capturing distinct genesets. Our results indicate that GWAS and EWAS are capturing different aspects of the biology of complex traits.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
