

In silico identification of multiple conserved motifs within the control region of Culicidae mitogenomes
- PMID: 36536037
- PMCID: PMC9763401
- DOI: 10.1038/s41598-022-26236-5
In silico identification of multiple conserved motifs within the control region of Culicidae mitogenomes
Abstract
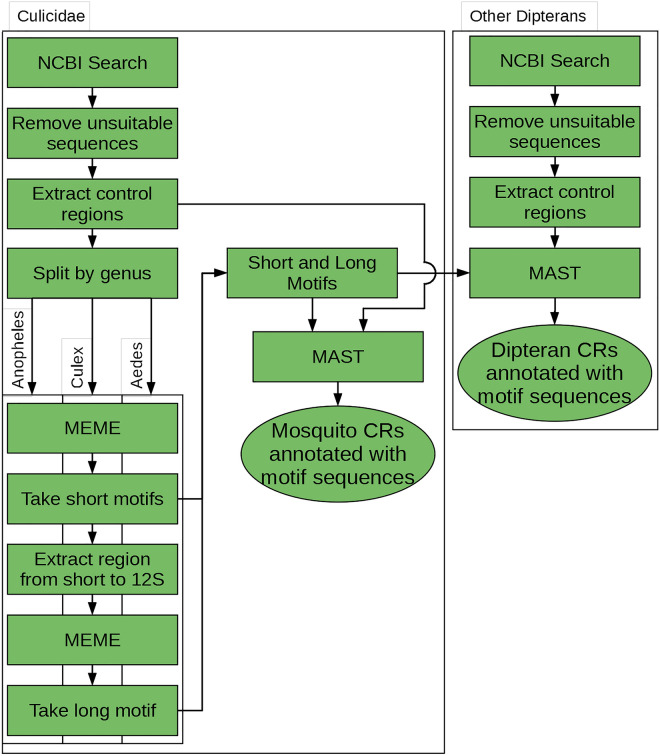
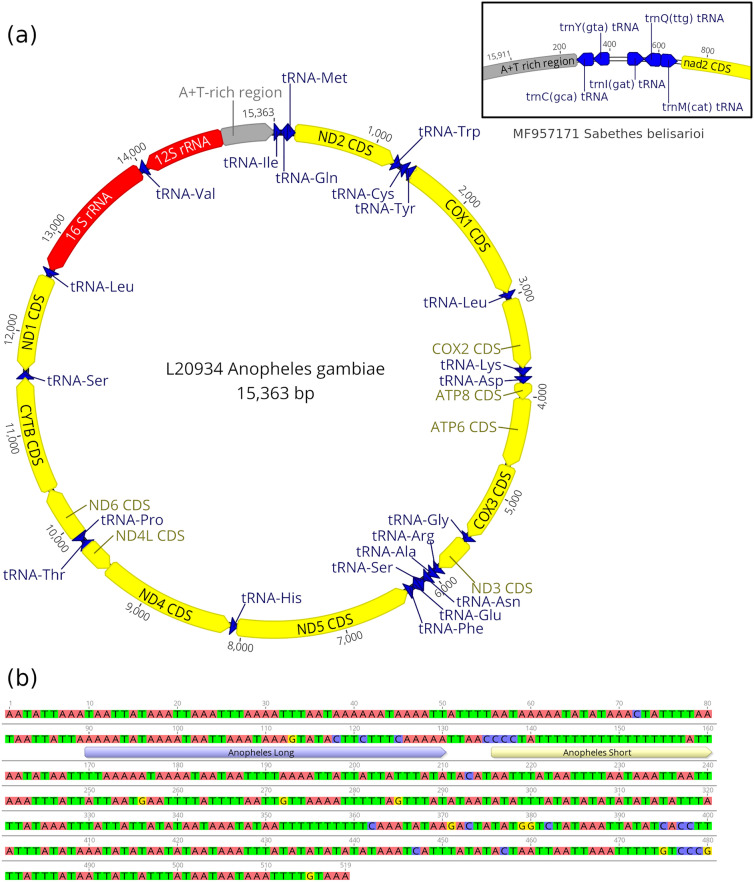
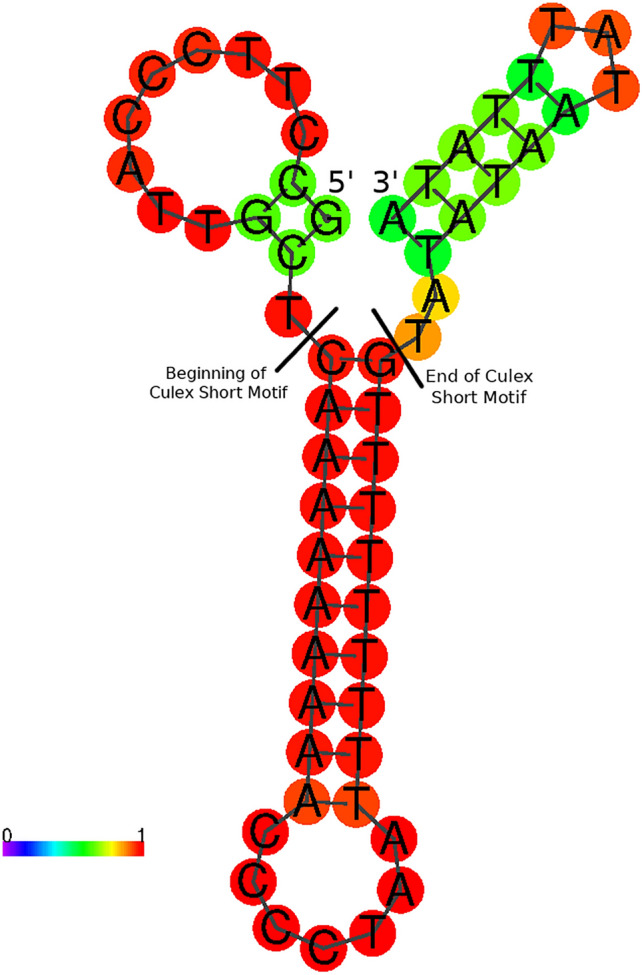
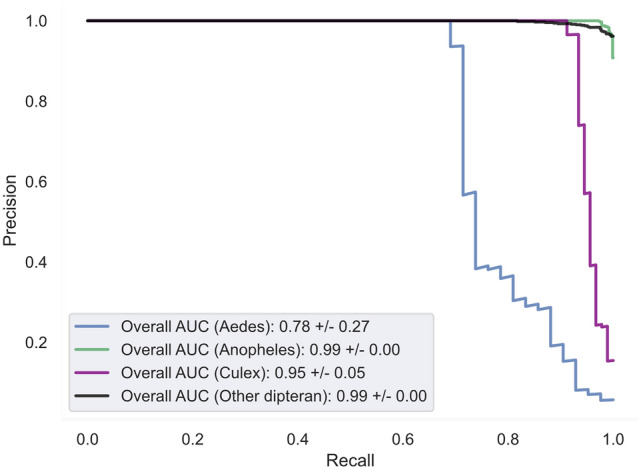
Mosquitoes are important vectors for human and animal diseases. Genetic markers, like the mitochondrial COI gene, can facilitate the taxonomic classification of disease vectors, vector-borne disease surveillance, and prevention. Within the control region (CR) of the mitochondrial genome, there exists a highly variable and poorly studied non-coding AT-rich area that contains the origin of replication. Although the CR hypervariable region has been used for species differentiation of some animals, few studies have investigated the mosquito CR. In this study, we analyze the mosquito mitogenome CR sequences from 125 species and 17 genera. We discovered four conserved motifs located 80 to 230 bp upstream of the 12S rRNA gene. Two of these motifs were found within all 392 Anopheles (An.) CR sequences while the other two motifs were identified in all 37 Culex (Cx.) CR sequences. However, only 3 of the 304 non-Culicidae Dipteran mitogenome CR sequences contained these motifs. Interestingly, the short motif found in all 37 Culex sequences had poly-A and poly-T stretch of similar length that is predicted to form a stable hairpin. We show that supervised learning using the frequency chaos game representation of the CR can be used to differentiate mosquito genera from their dipteran relatives.
© 2022. Crown.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- World Malaria Report 2019. Available from: https://www.who.int/publications-detail-redirect/9789241565721 (2020).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
