

Nano3P-seq: transcriptome-wide analysis of gene expression and tail dynamics using end-capture nanopore cDNA sequencing
- PMID: 36536091
- PMCID: PMC9834059
- DOI: 10.1038/s41592-022-01714-w
Nano3P-seq: transcriptome-wide analysis of gene expression and tail dynamics using end-capture nanopore cDNA sequencing
Abstract
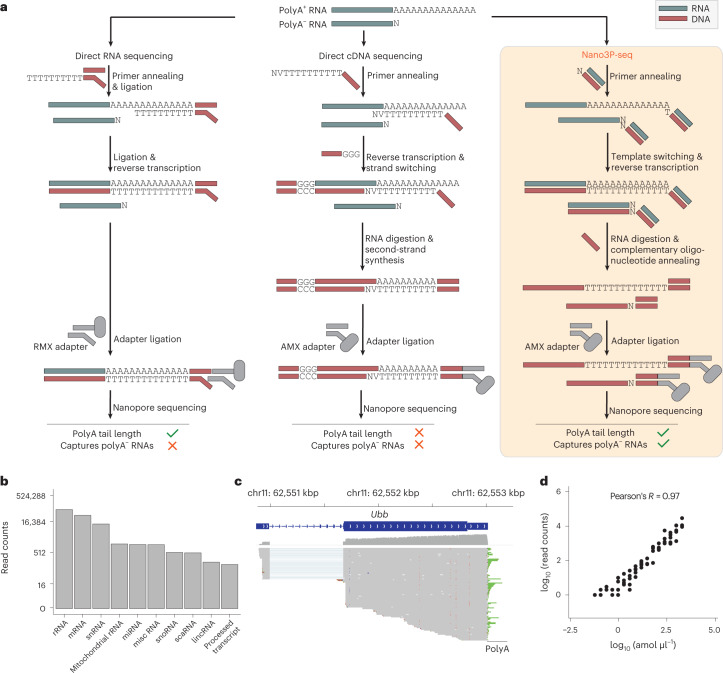
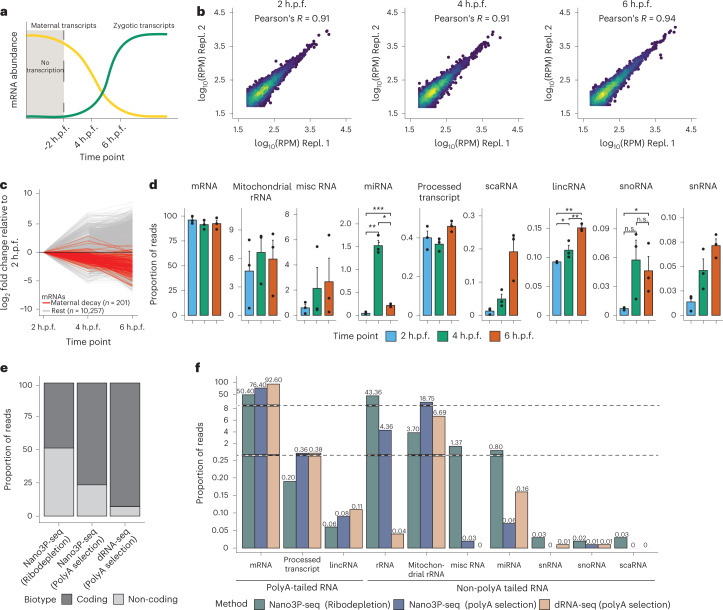
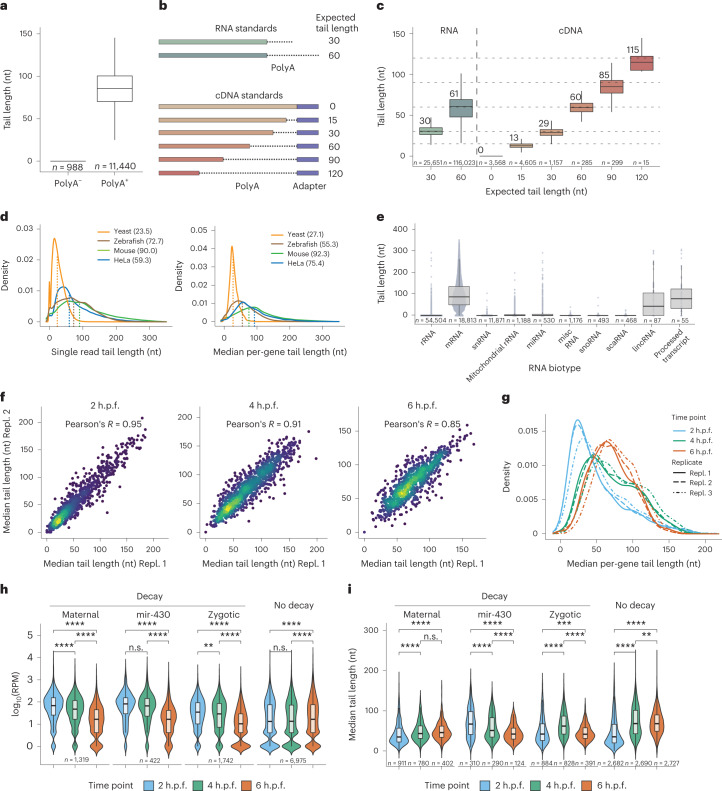
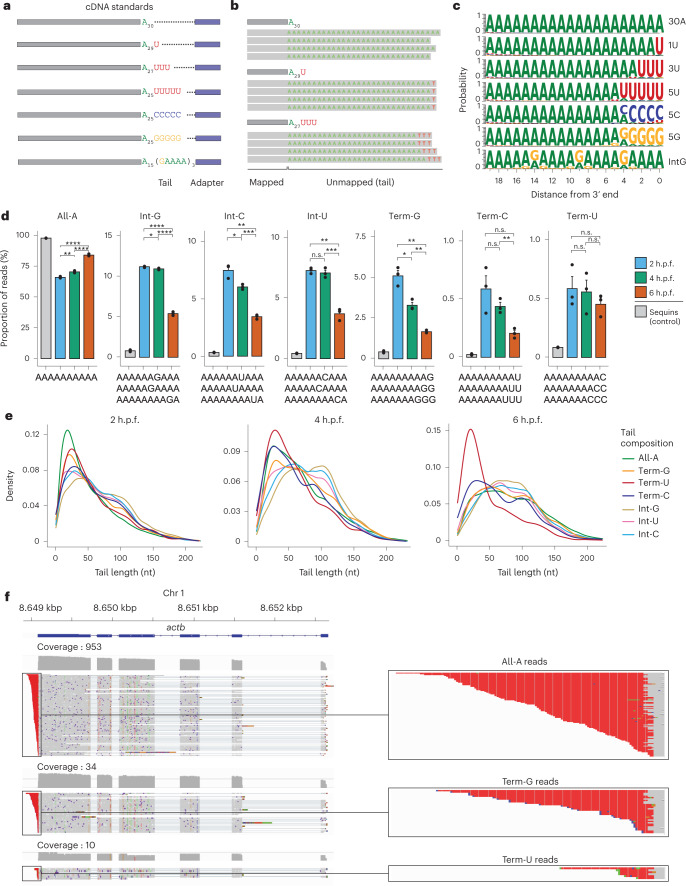
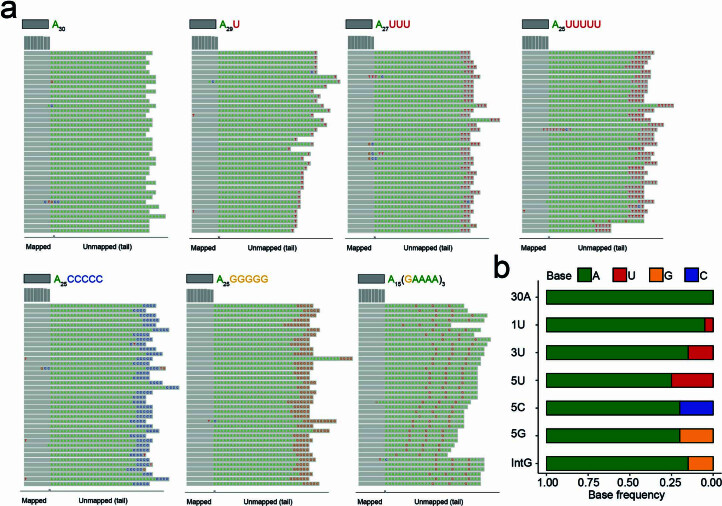
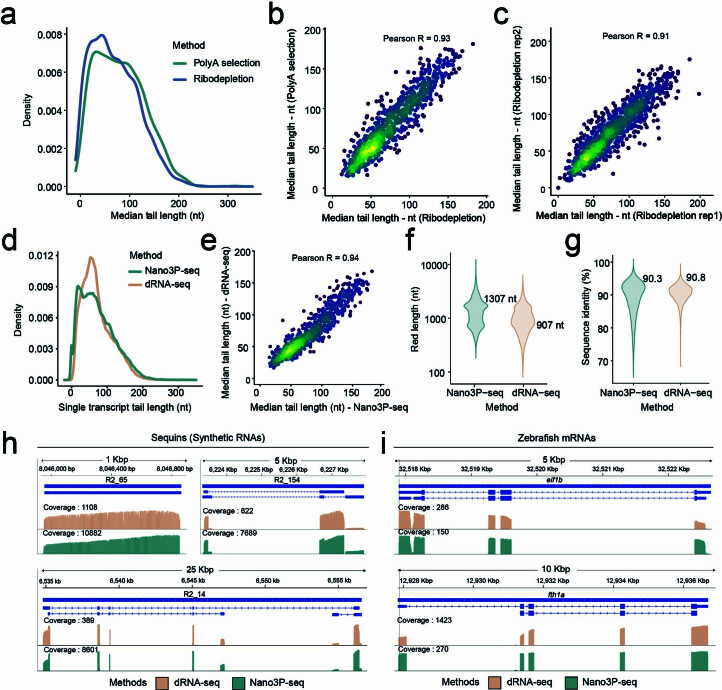
RNA polyadenylation plays a central role in RNA maturation, fate, and stability. In response to developmental cues, polyA tail lengths can vary, affecting the translation efficiency and stability of mRNAs. Here we develop Nanopore 3' end-capture sequencing (Nano3P-seq), a method that relies on nanopore cDNA sequencing to simultaneously quantify RNA abundance, tail composition, and tail length dynamics at per-read resolution. By employing a template-switching-based sequencing protocol, Nano3P-seq can sequence RNA molecule from its 3' end, regardless of its polyadenylation status, without the need for PCR amplification or ligation of RNA adapters. We demonstrate that Nano3P-seq provides quantitative estimates of RNA abundance and tail lengths, and captures a wide diversity of RNA biotypes. We find that, in addition to mRNA and long non-coding RNA, polyA tails can be identified in 16S mitochondrial ribosomal RNA in both mouse and zebrafish models. Moreover, we show that mRNA tail lengths are dynamically regulated during vertebrate embryogenesis at an isoform-specific level, correlating with mRNA decay. Finally, we demonstrate the ability of Nano3P-seq in capturing non-A bases within polyA tails of various lengths, and reveal their distribution during vertebrate embryogenesis. Overall, Nano3P-seq is a simple and robust method for accurately estimating transcript levels, tail lengths, and tail composition heterogeneity in individual reads, with minimal library preparation biases, both in the coding and non-coding transcriptome.
© 2022. The Author(s).
Conflict of interest statement
E.M.N. has received travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences. E.M.N. is a member of the Scientific Advisory Board of IMMAGINA Biotech. All authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures








References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
