

Profound phenotypic and epigenetic heterogeneity of the HIV-1-infected CD4+ T cell reservoir
- PMID: 36536105
- PMCID: PMC9892009
- DOI: 10.1038/s41590-022-01371-3
Profound phenotypic and epigenetic heterogeneity of the HIV-1-infected CD4+ T cell reservoir
Abstract
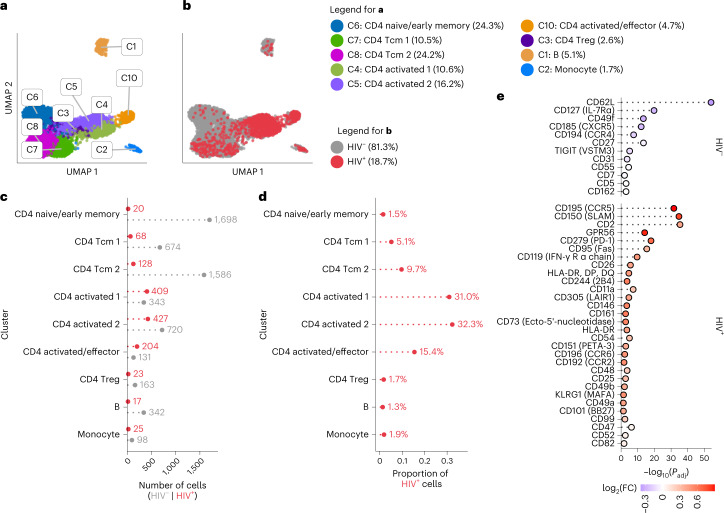
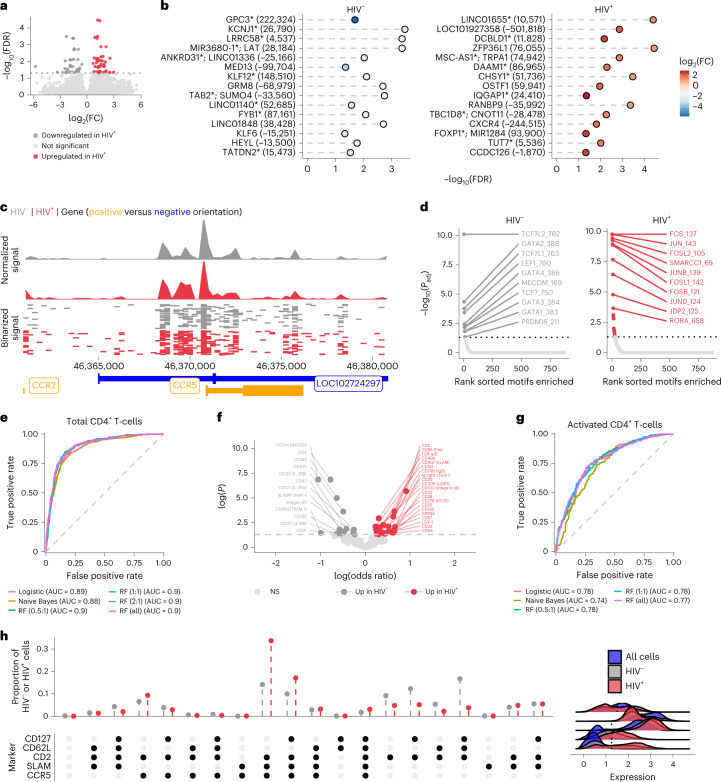
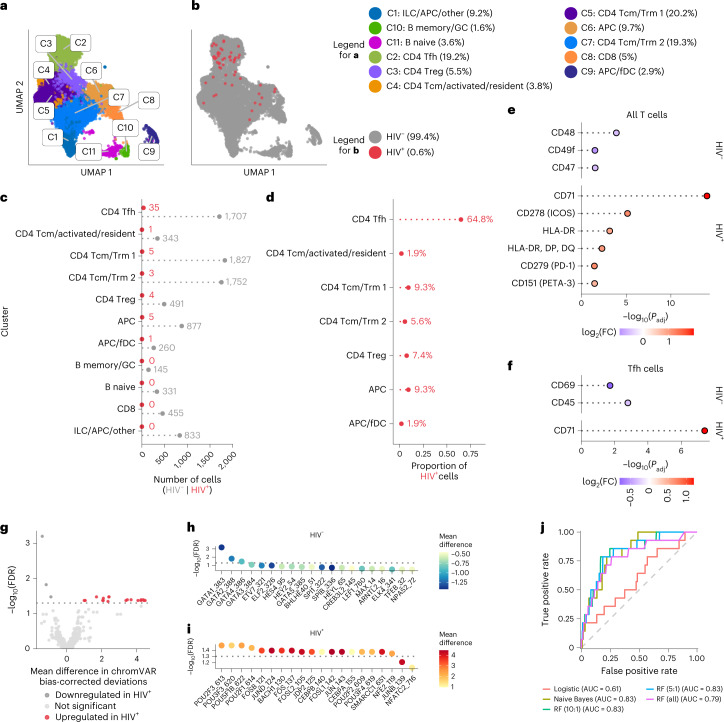
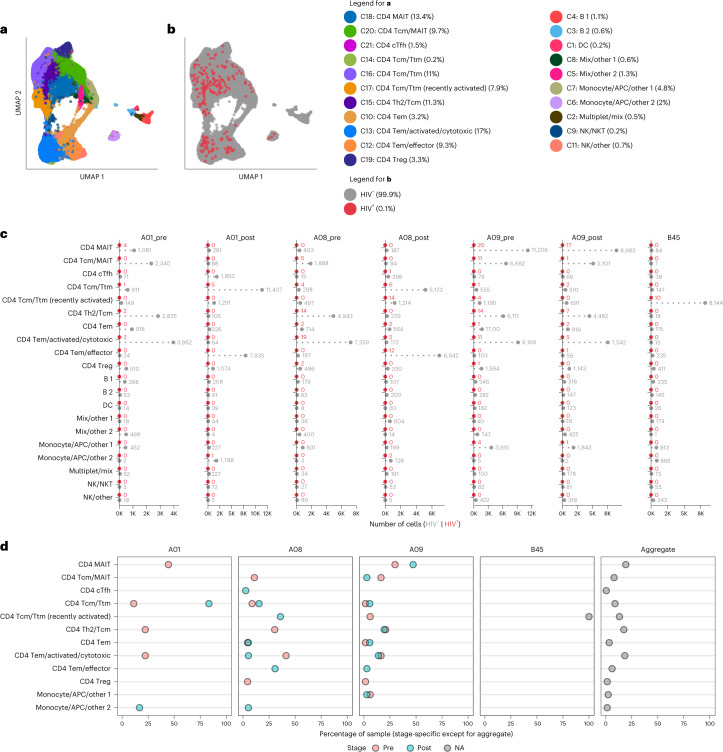
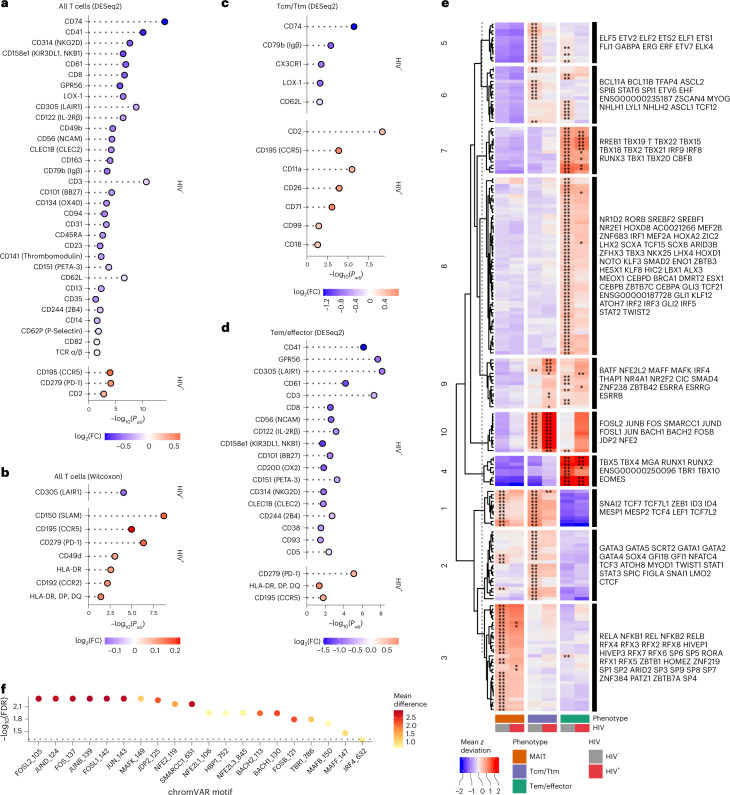
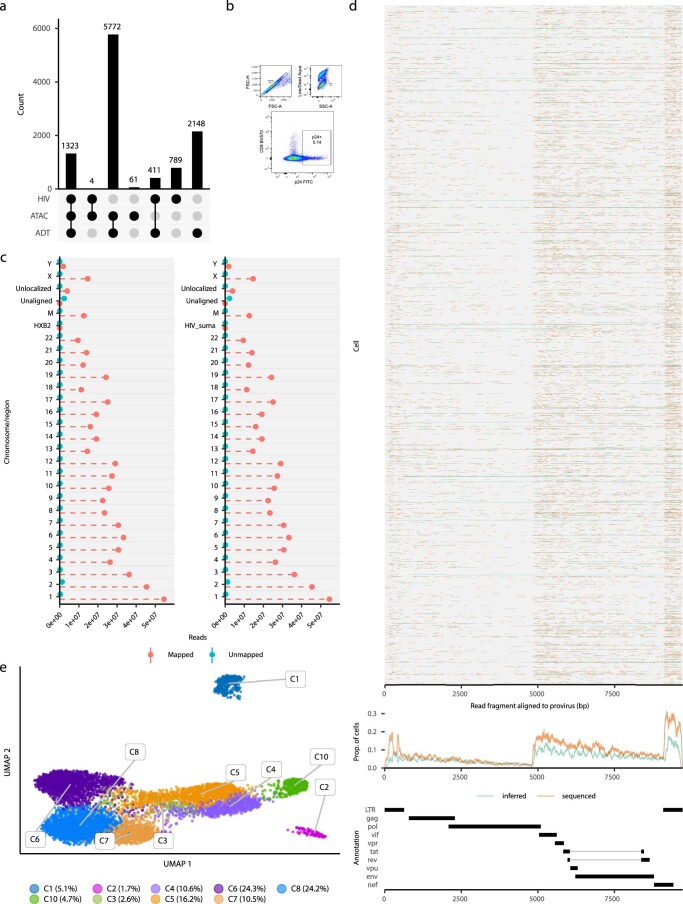
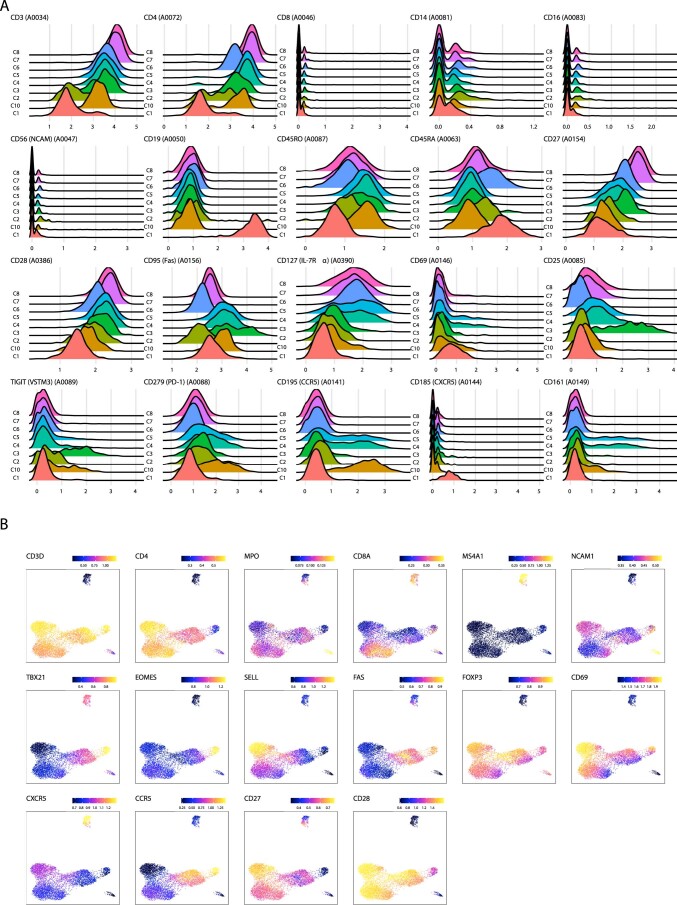
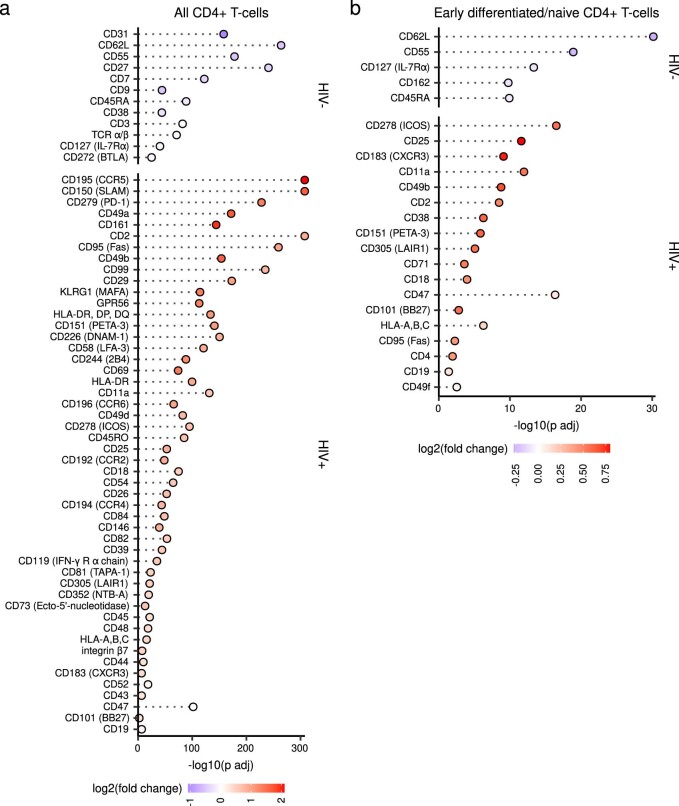
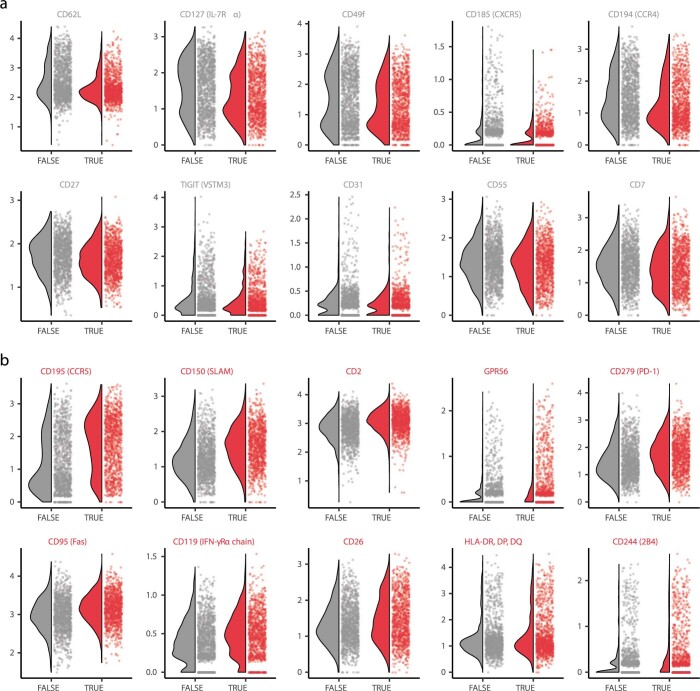
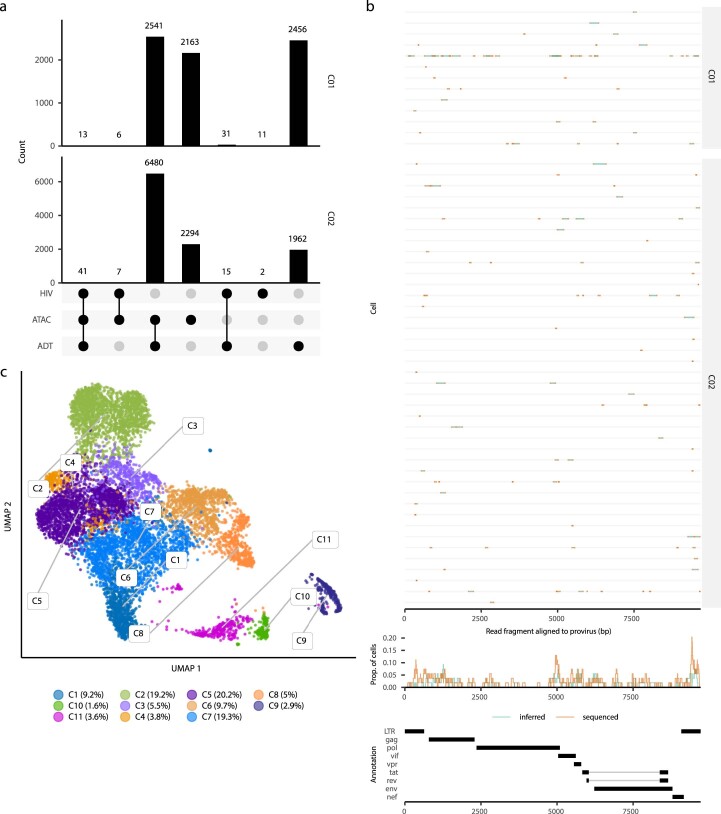
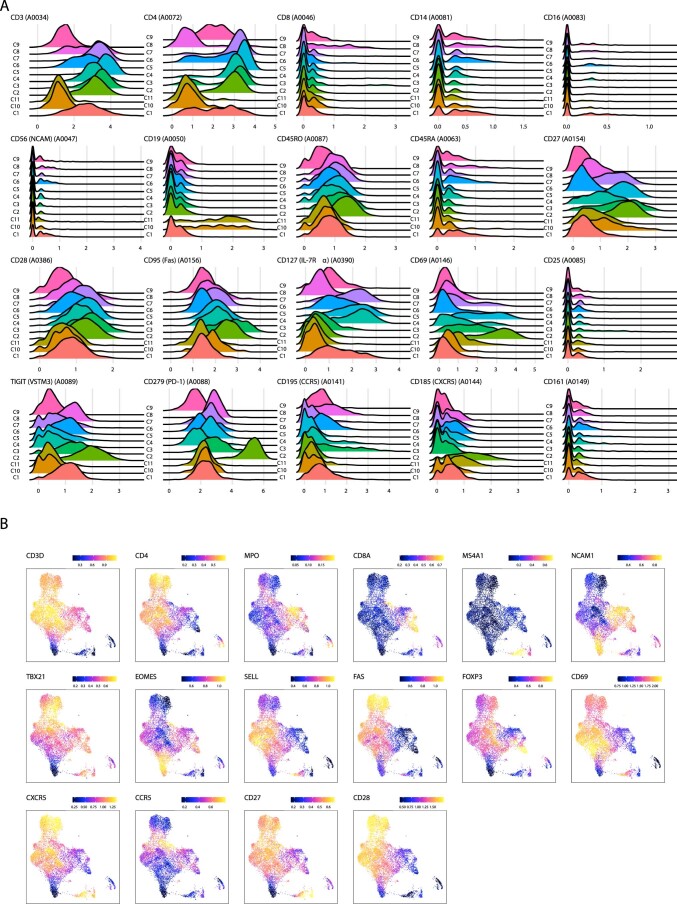
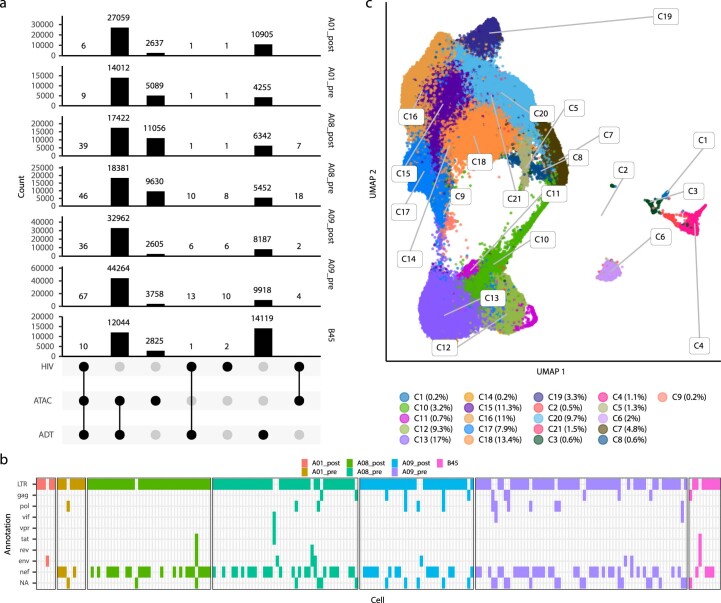
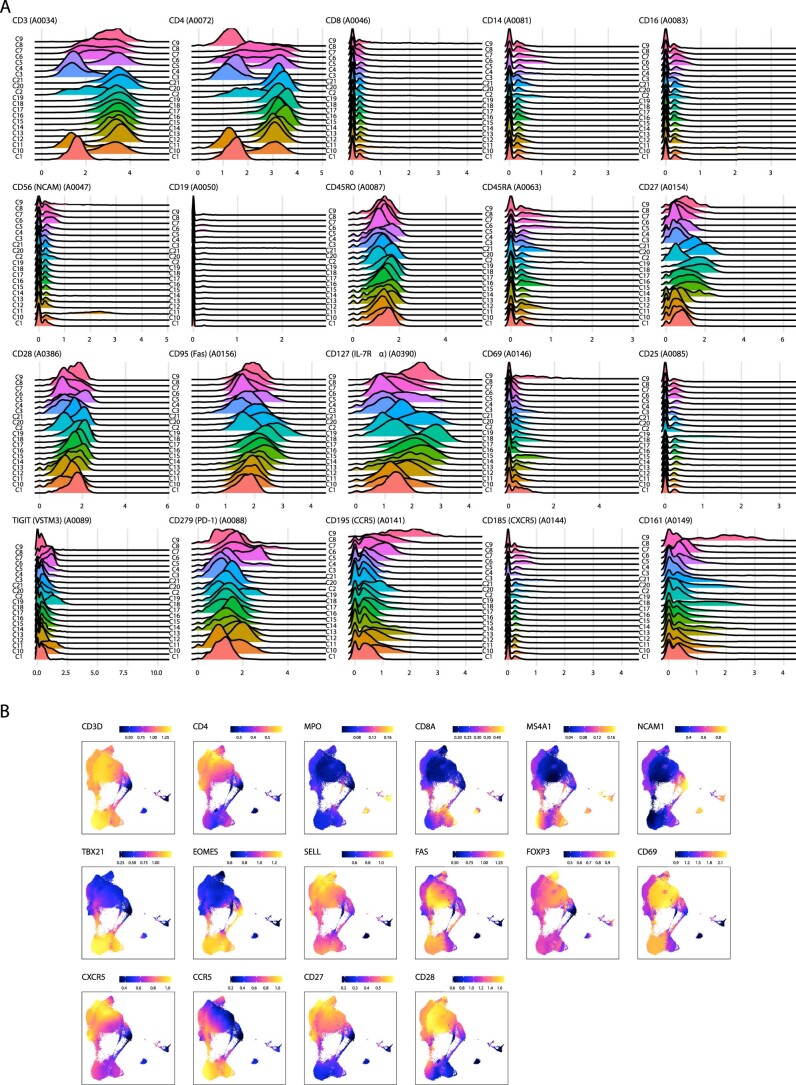
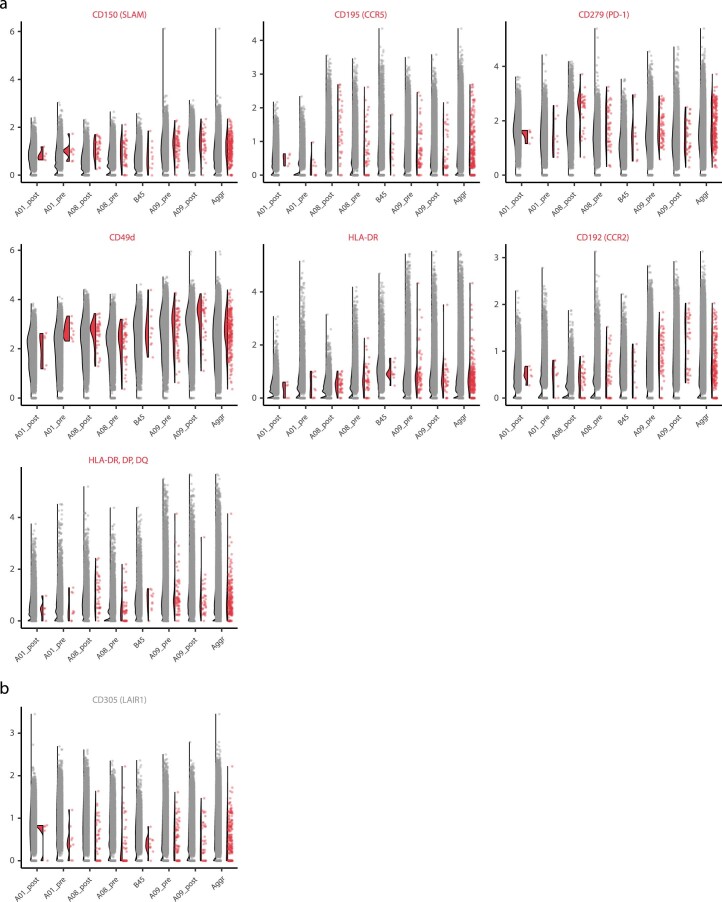
Understanding the complexity of the long-lived HIV reservoir during antiretroviral therapy (ART) remains a considerable impediment in research towards a cure for HIV. To address this, we developed a single-cell strategy to precisely define the unperturbed peripheral blood HIV-infected memory CD4+ T cell reservoir from ART-treated people living with HIV (ART-PLWH) via the presence of integrated accessible proviral DNA in concert with epigenetic and cell surface protein profiling. We identified profound reservoir heterogeneity within and between ART-PLWH, characterized by new and known surface markers within total and individual memory CD4+ T cell subsets. We further uncovered new epigenetic profiles and transcription factor motifs enriched in HIV-infected cells that suggest infected cells with accessible provirus, irrespective of reservoir distribution, are poised for reactivation during ART treatment. Together, our findings reveal the extensive inter- and intrapersonal cellular heterogeneity of the HIV reservoir, and establish an initial multiomic atlas to develop targeted reservoir elimination strategies.
© 2022. The Author(s).
Conflict of interest statement
M.R.B. is a consultant for Interius BioTherapeutics. No other conflicts are reported by the authors.
Figures
References
-
- Chun TW, et al. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1995;1:1284–1290. - PubMed
-
- Finzi D, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. - PubMed
-
- Chun TW, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
