

Loss of G0/G1 switch gene 2 (G0S2) promotes disease progression and drug resistance in chronic myeloid leukaemia (CML) by disrupting glycerophospholipid metabolism
- PMID: 36536477
- PMCID: PMC9763536
- DOI: 10.1002/ctm2.1146
Loss of G0/G1 switch gene 2 (G0S2) promotes disease progression and drug resistance in chronic myeloid leukaemia (CML) by disrupting glycerophospholipid metabolism
Abstract
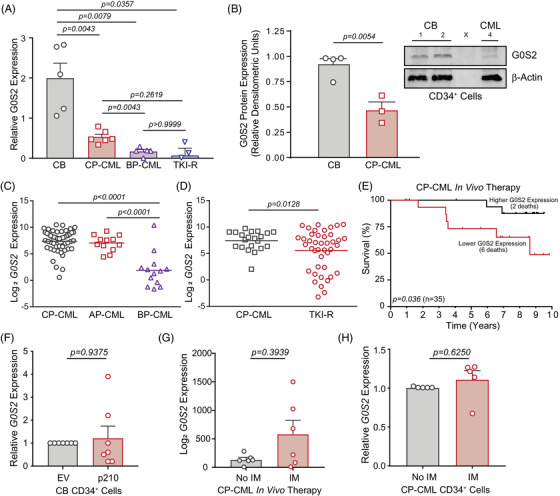
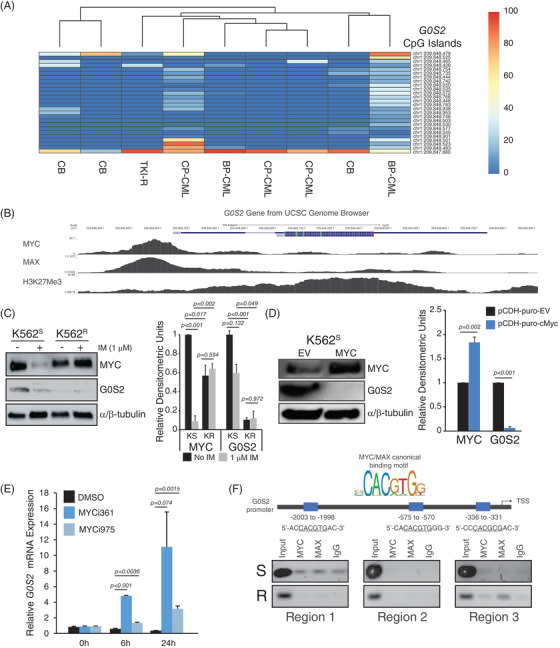
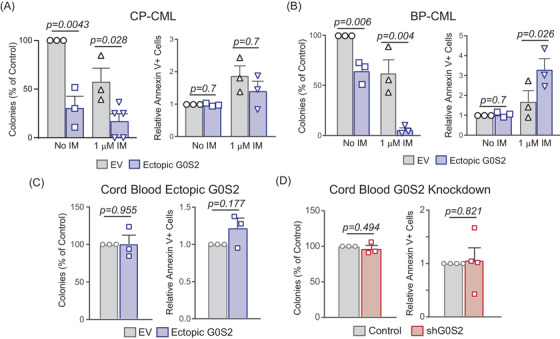
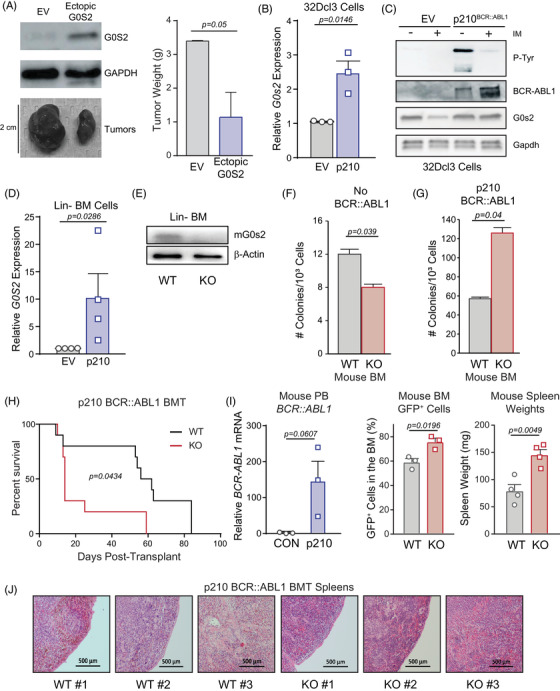
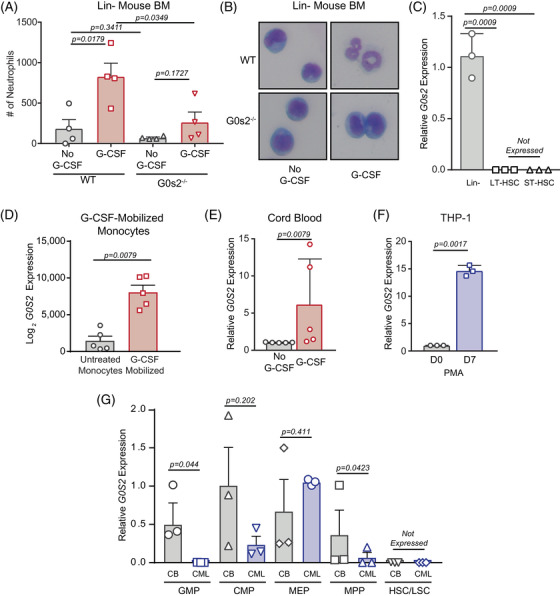
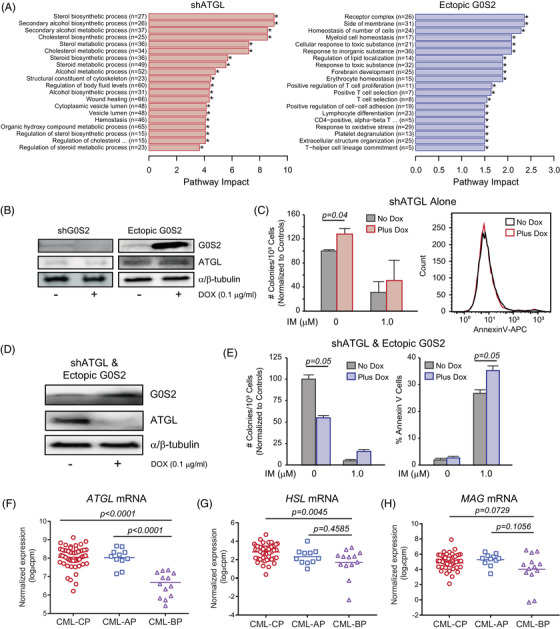
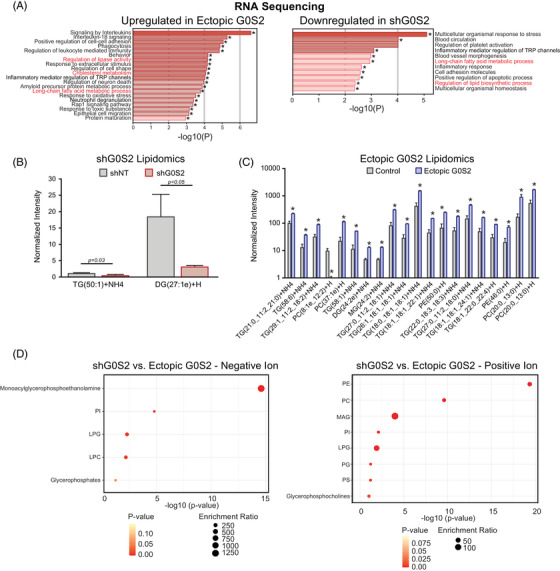
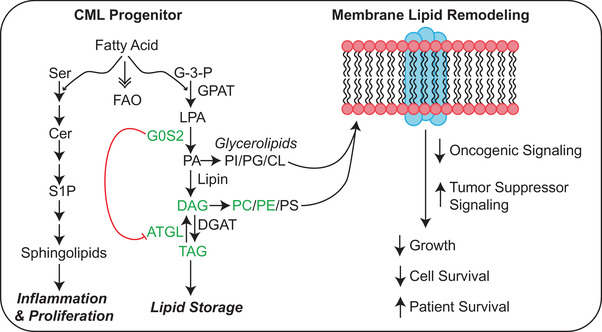
Tyrosine kinase inhibitors (TKIs) targeting BCR::ABL1 have turned chronic myeloid leukaemia (CML) from a fatal disease into a manageable condition for most patients. Despite improved survival, targeting drug-resistant leukaemia stem cells (LSCs) remains a challenge for curative CML therapy. Aberrant lipid metabolism can have a large impact on membrane dynamics, cell survival and therapeutic responses in cancer. While ceramide and sphingolipid levels were previously correlated with TKI response in CML, the role of lipid metabolism in TKI resistance is not well understood. We have identified downregulation of a critical regulator of lipid metabolism, G0/G1 switch gene 2 (G0S2), in multiple scenarios of TKI resistance, including (1) BCR::ABL1 kinase-independent TKI resistance, (2) progression of CML from the chronic to the blast phase of the disease, and (3) in CML versus normal myeloid progenitors. Accordingly, CML patients with low G0S2 expression levels had a worse overall survival. G0S2 downregulation in CML was not a result of promoter hypermethylation or BCR::ABL1 kinase activity, but was rather due to transcriptional repression by MYC. Using CML cell lines, patient samples and G0s2 knockout (G0s2-/- ) mice, we demonstrate a tumour suppressor role for G0S2 in CML and TKI resistance. Our data suggest that reduced G0S2 protein expression in CML disrupts glycerophospholipid metabolism, correlating with a block of differentiation that renders CML cells resistant to therapy. Altogether, our data unravel a new role for G0S2 in regulating myeloid differentiation and TKI response in CML, and suggest that restoring G0S2 may have clinical utility.
Keywords: G0/G1 switch gene 2 (G0S2); chronic myeloid leukaemia (CML); glycerophospholipid metabolism; tyrosine kinase inhibitor (TKI) resistance.
© 2022 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
B.J.D. serves on scientific advisory boards for Aileron Therapeutics, Therapy Architects (ALLCRON), Cepheid, Vivid Biosciences, Celgene, RUNX1 Research Program, Novartis, Gilead Sciences (inactive), Monojul (inactive); serves on Scientific Advisory Boards and receives stock from Aptose Biosciences, Blueprint Medicines, EnLiven Therapeutics, Iterion Therapeutics, Third Coast Therapeutics, GRAIL (inactive on scientific advisory board); is scientific founder of MolecularMD (inactive, acquired by ICON); serves on the board of directors and receives stock from Amgen, Vincera Pharma; serves on the board of directors for Burroughs Wellcome Fund, CureOne; serves on the joint steering committee for Beat AML LLS; is founder of VB Therapeutics; has a sponsored research agreement with EnLiven Therapeutics; receives clinical trial funding from Novartis, Bristol‐Myers Squibb, Pfizer. The remaining authors have no competing financial interests.
Figures
References
-
- Nowell PC, Hungerford D. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497. - PubMed
-
- Druker BJ, Guilhot F, O'Brien SG, et al. Five‐year follow‐up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408‐2417. - PubMed
-
- Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103(11):4010‐4022. - PubMed
-
- O'Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12(8):513‐526. - PubMed
-
- Graham SM, Jorgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia‐positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319‐325. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous