

Improved genome editing by an engineered CRISPR-Cas12a
- PMID: 36537251
- PMCID: PMC9825149
- DOI: 10.1093/nar/gkac1192
Improved genome editing by an engineered CRISPR-Cas12a
Abstract
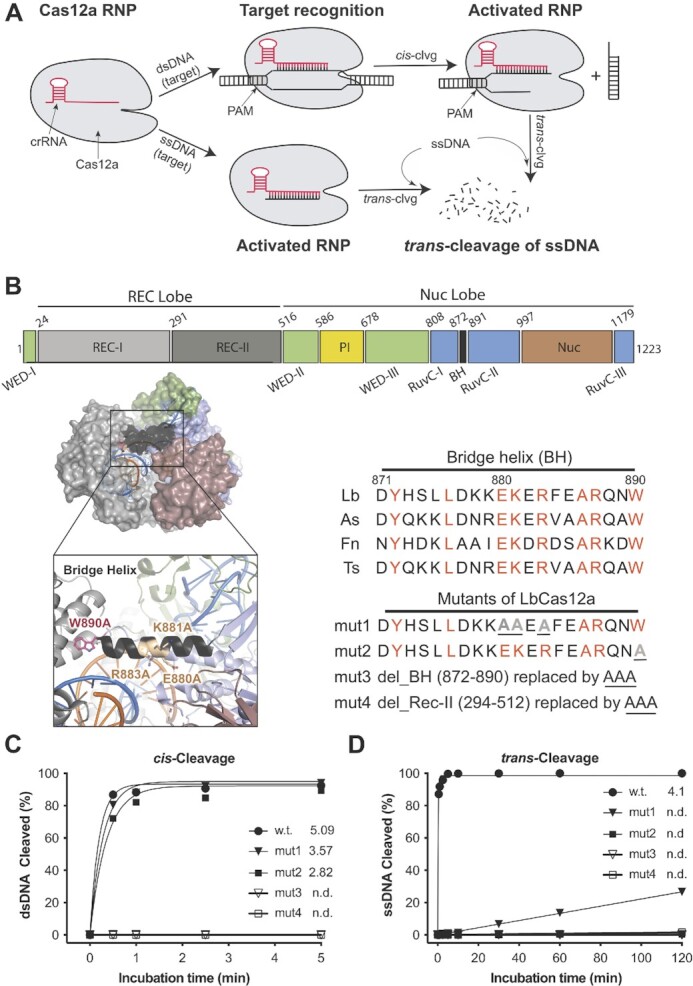
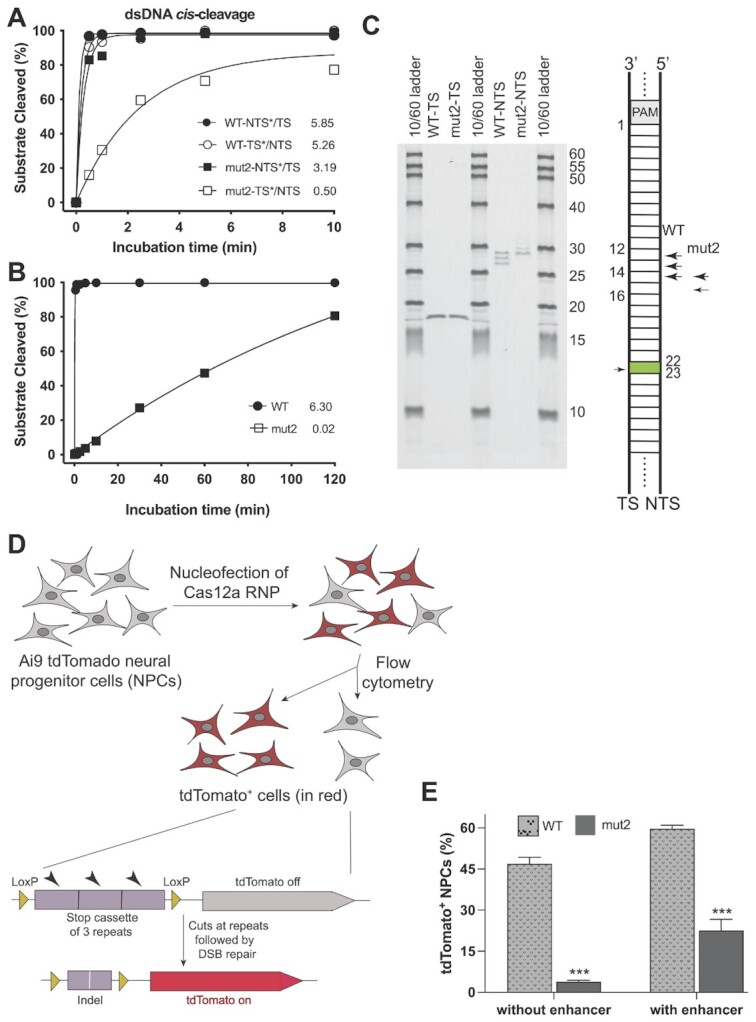
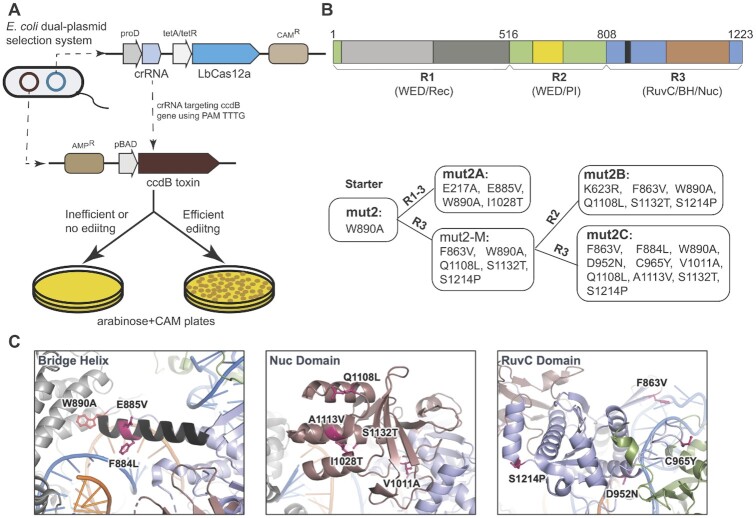
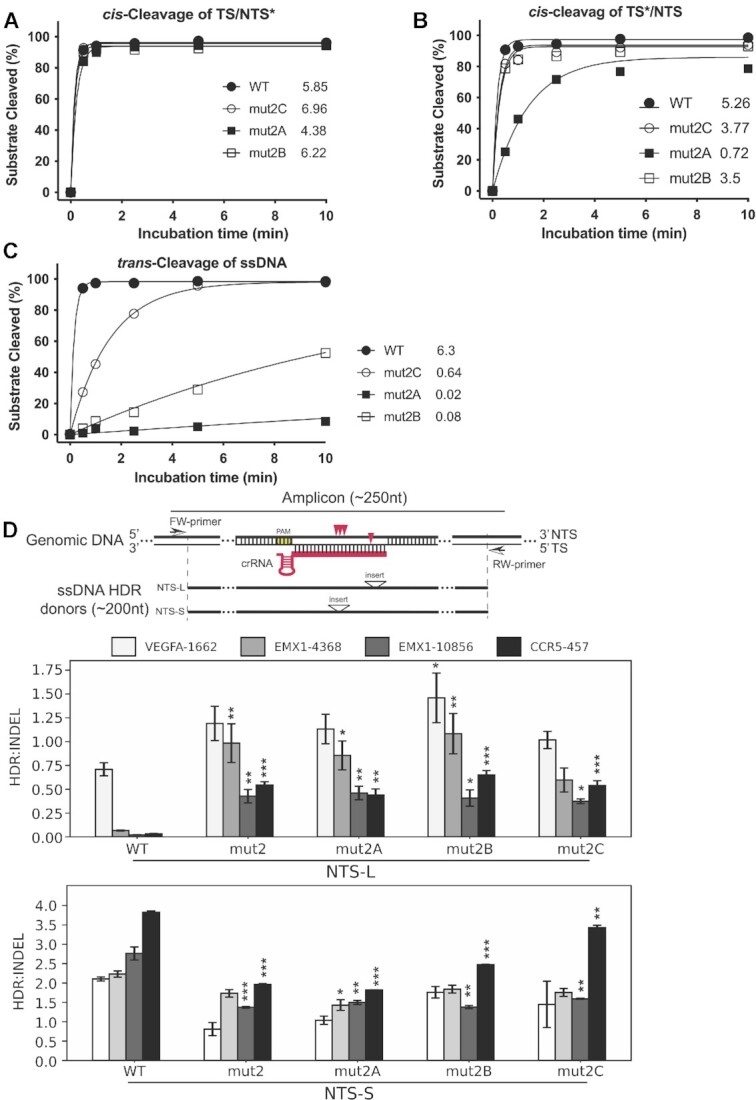
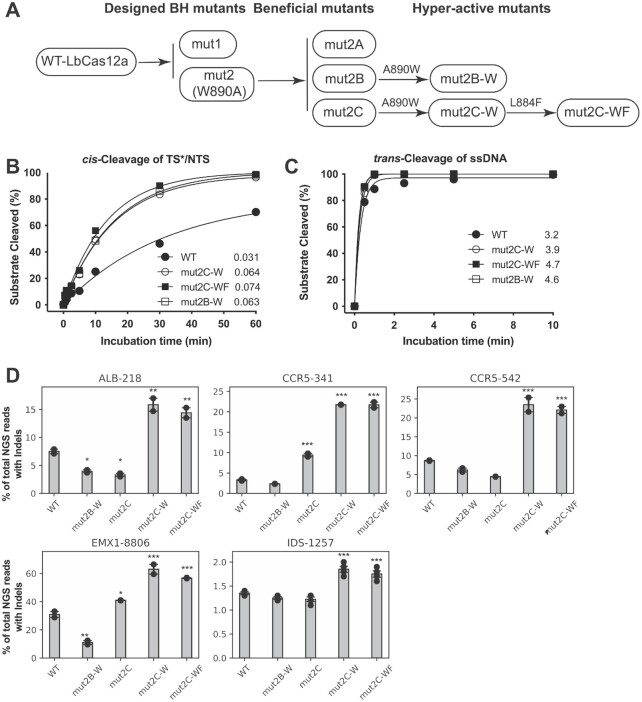
CRISPR-Cas12a is an RNA-guided, programmable genome editing enzyme found within bacterial adaptive immune pathways. Unlike CRISPR-Cas9, Cas12a uses only a single catalytic site to both cleave target double-stranded DNA (dsDNA) (cis-activity) and indiscriminately degrade single-stranded DNA (ssDNA) (trans-activity). To investigate how the relative potency of cis- versus trans-DNase activity affects Cas12a-mediated genome editing, we first used structure-guided engineering to generate variants of Lachnospiraceae bacterium Cas12a that selectively disrupt trans-activity. The resulting engineered mutant with the biggest differential between cis- and trans-DNase activity in vitro showed minimal genome editing activity in human cells, motivating a second set of experiments using directed evolution to generate additional mutants with robust genome editing activity. Notably, these engineered and evolved mutants had enhanced ability to induce homology-directed repair (HDR) editing by 2-18-fold compared to wild-type Cas12a when using HDR donors containing mismatches with crRNA at the PAM-distal region. Finally, a site-specific reversion mutation produced improved Cas12a (iCas12a) variants with superior genome editing efficiency at genomic sites that are difficult to edit using wild-type Cas12a. This strategy establishes a pipeline for creating improved genome editing tools by combining structural insights with randomization and selection. The available structures of other CRISPR-Cas enzymes will enable this strategy to be applied to improve the efficacy of other genome-editing proteins.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Jiang W., Marraffini L.A.. CRISPR-Cas: new tools for genetic manipulations from bacterial immunity systems. Annu. Rev. Microbiol. 2015; 69:209–228. - PubMed
-
- Wang J.Y., Pausch P., Doudna J.A.. Structural biology of CRISPR-Cas immunity and genome editing enzymes. Nat. Rev. Microbiol. 2022; 20:641–656. - PubMed
-
- Zhang F., Huang Z.. Mechanistic insights into the versatile class II CRISPR toolbox. Trends Biochem. Sci. 2022; 47:433–450. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
