

A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets
- PMID: 36539501
- PMCID: PMC9767870
- DOI: 10.1038/s43018-022-00475-x
A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets
Abstract
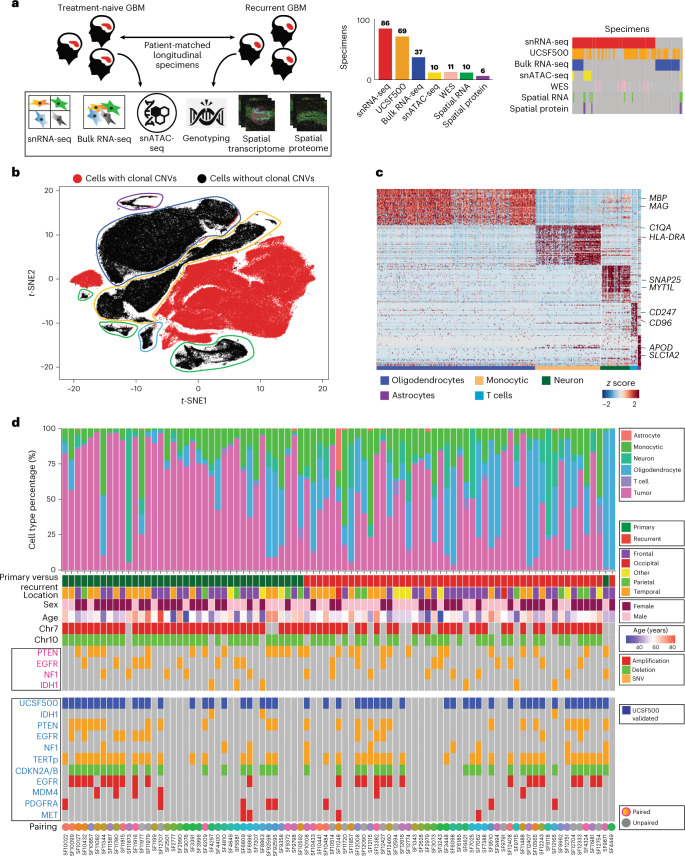
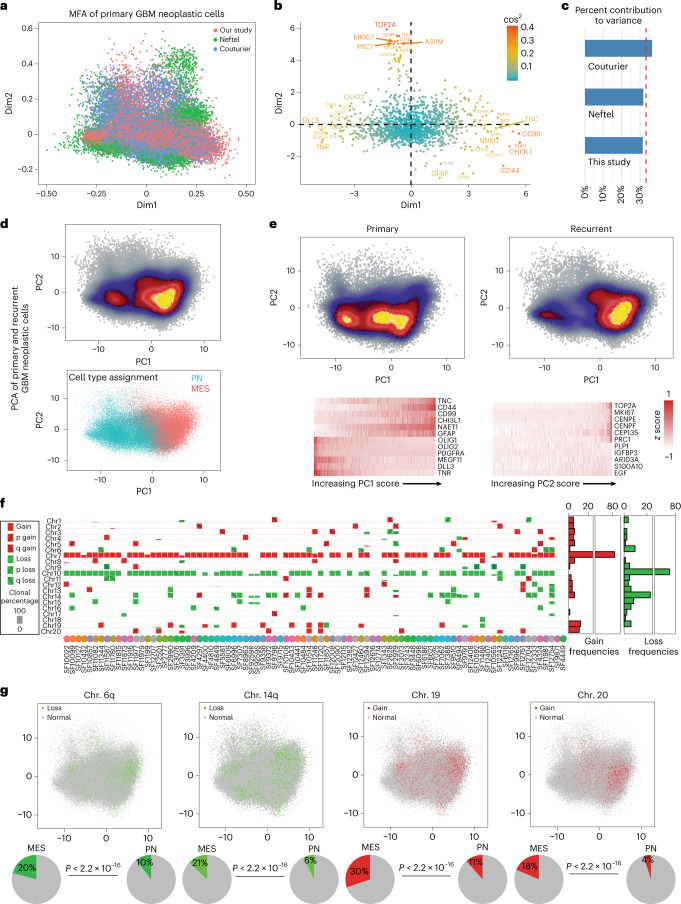
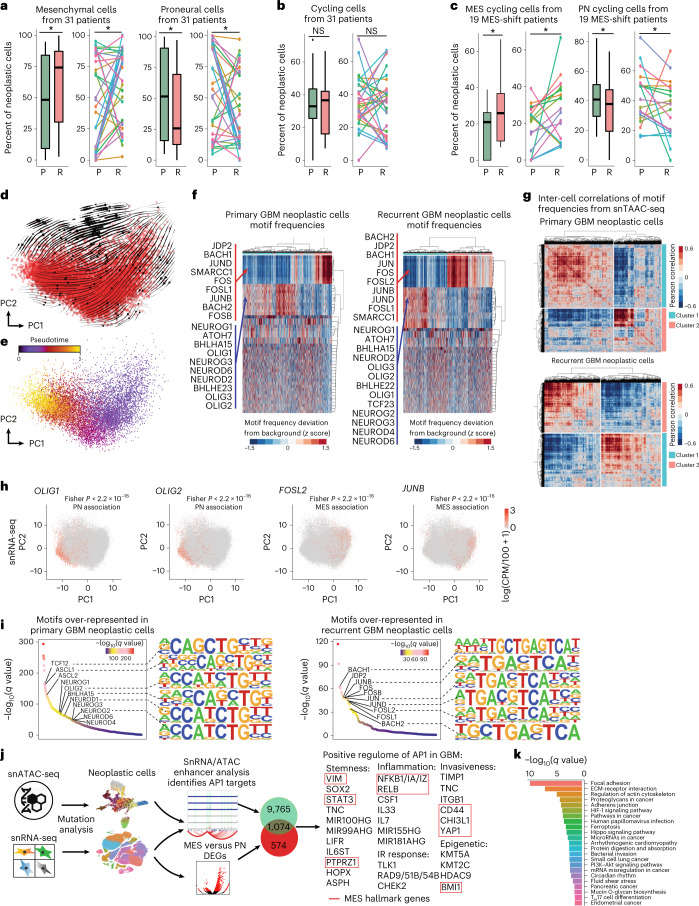
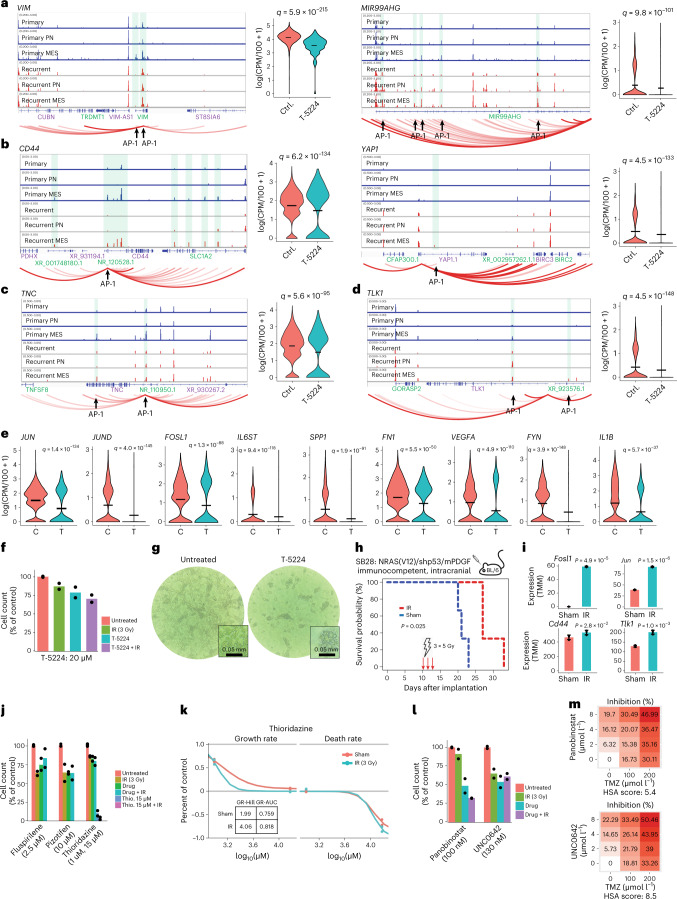
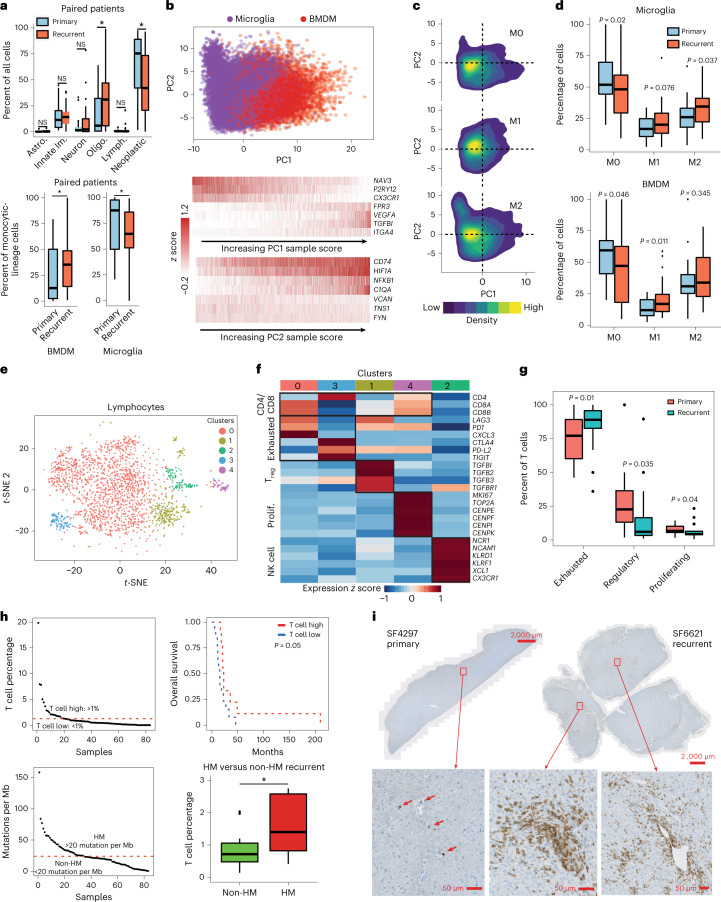
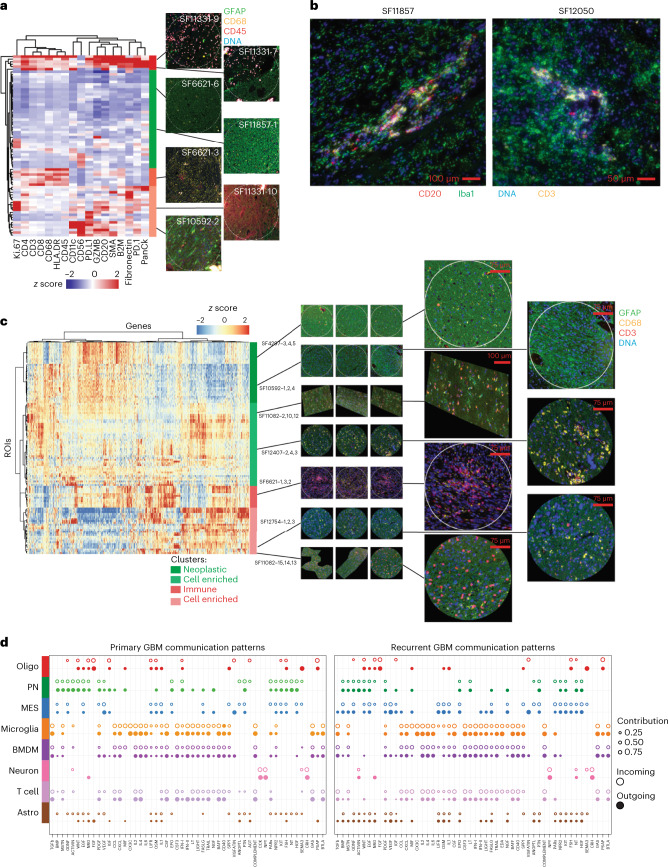
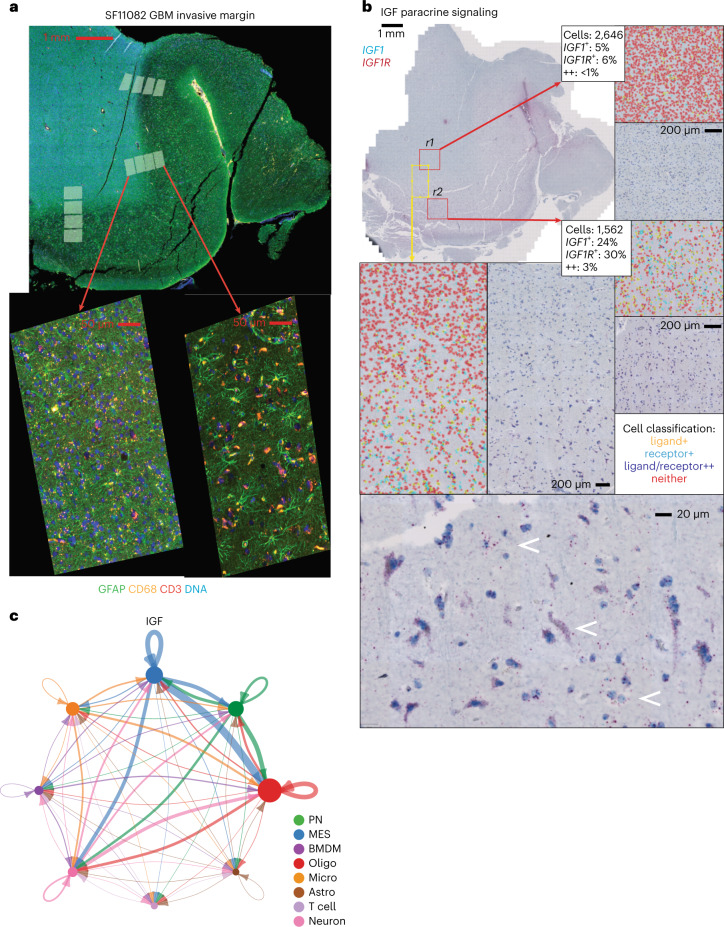
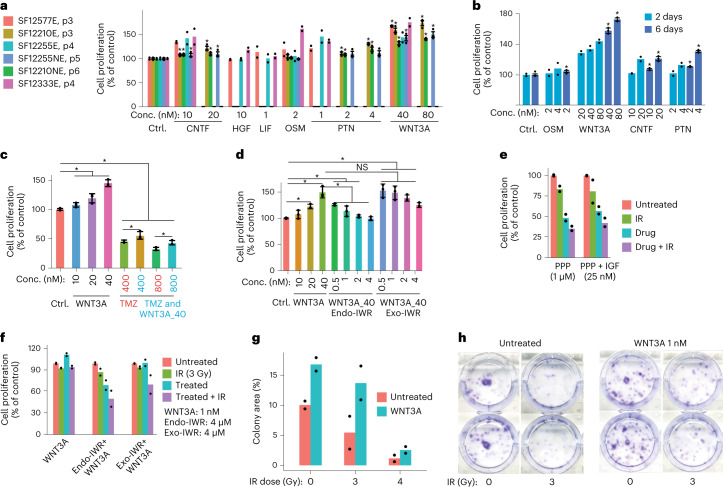
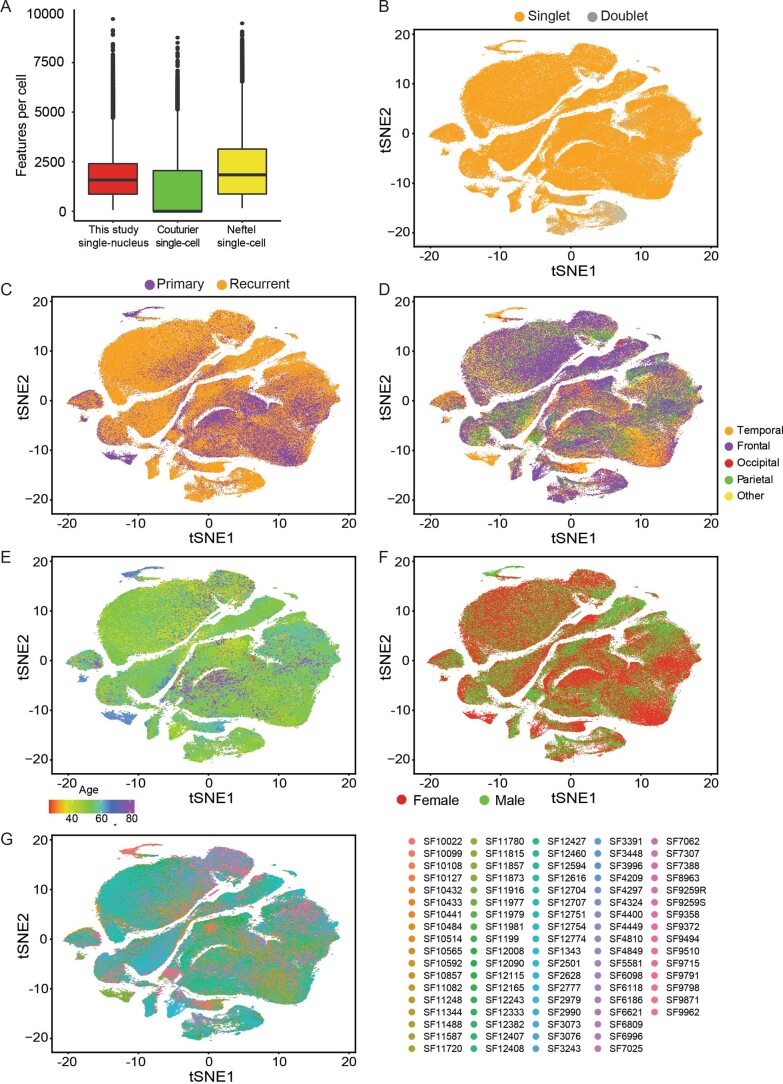
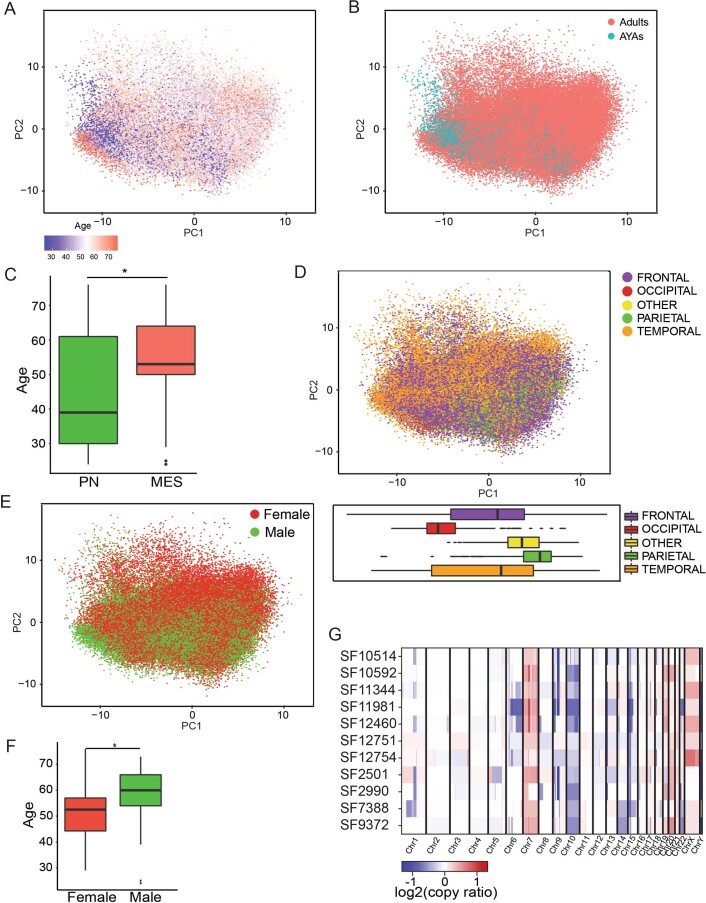
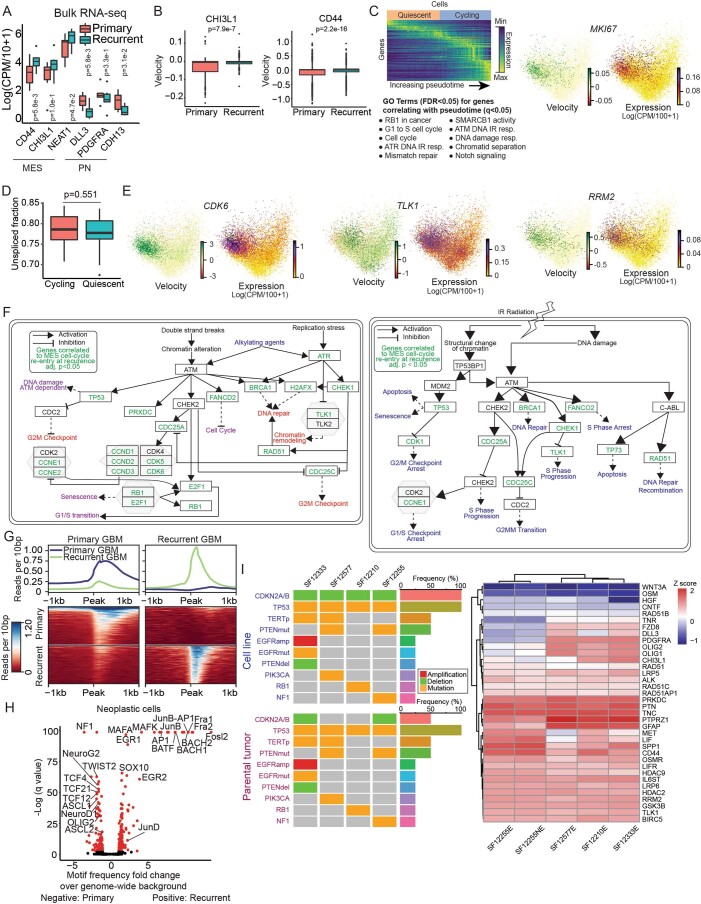
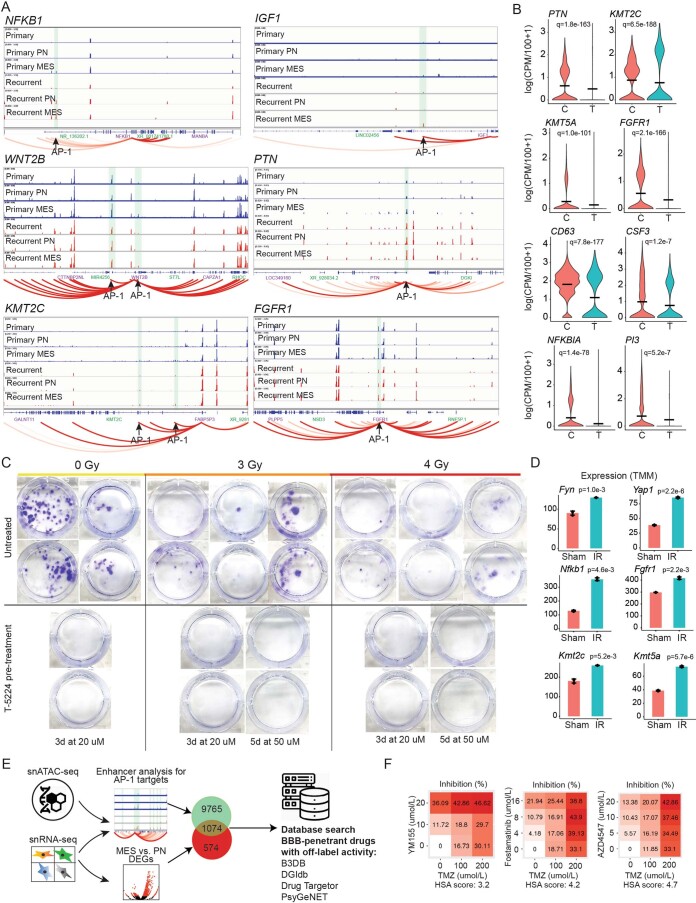
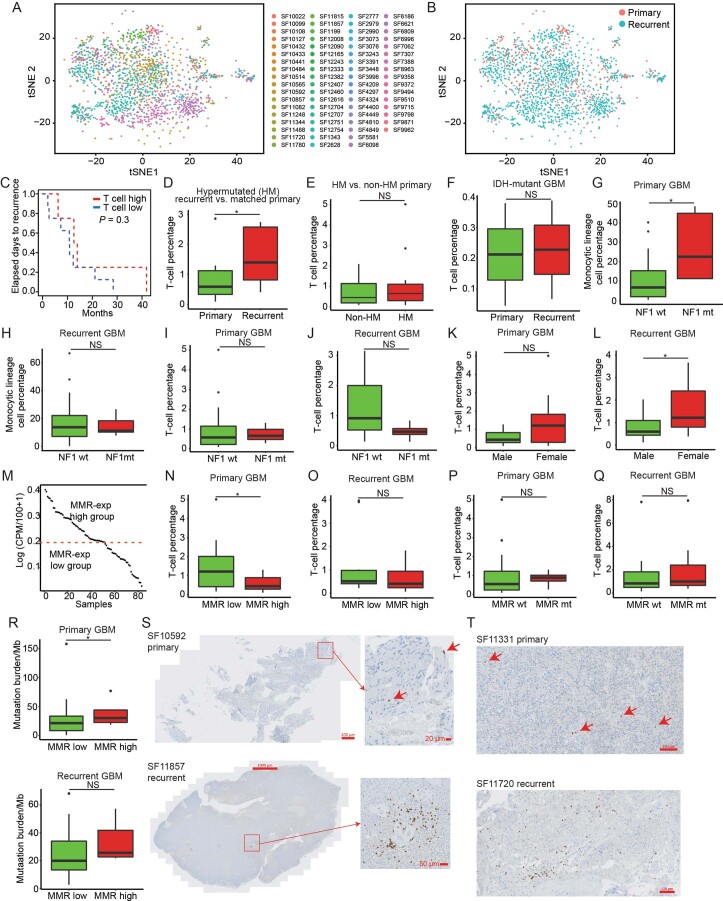
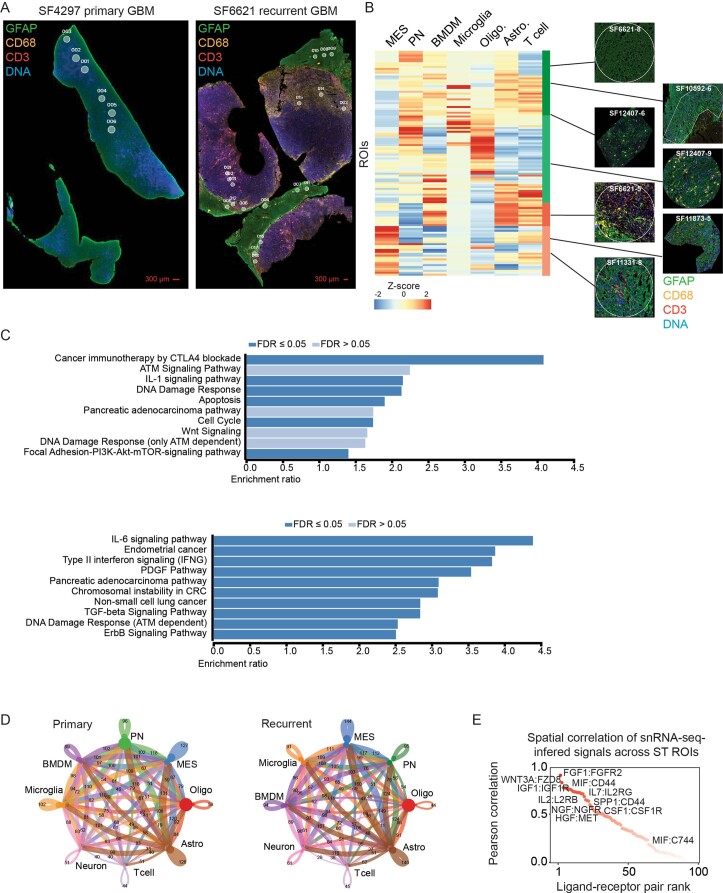
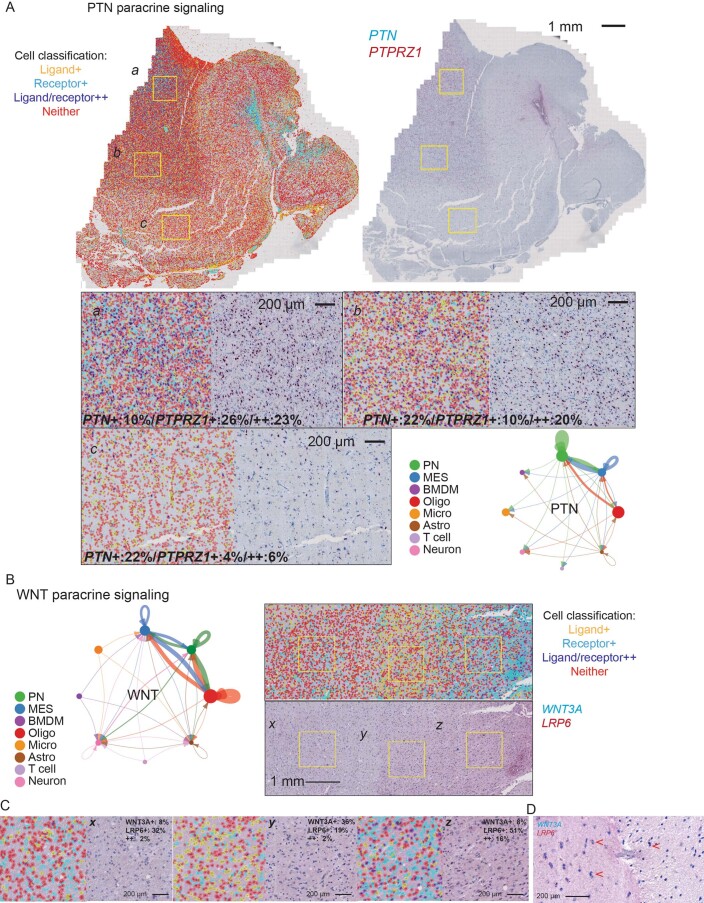
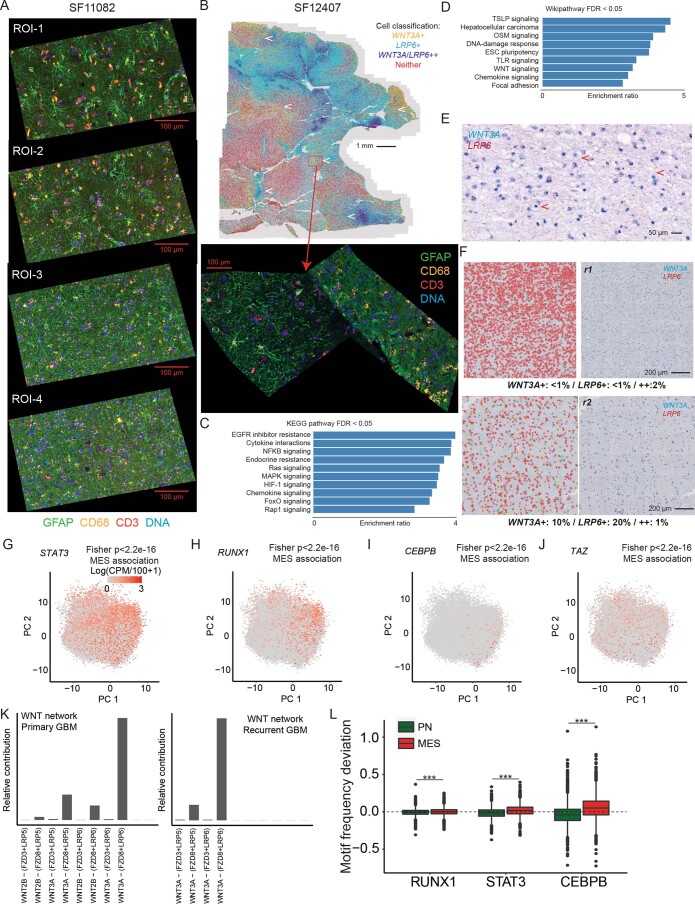
Recent longitudinal studies of glioblastoma (GBM) have demonstrated a lack of apparent selection pressure for specific DNA mutations in recurrent disease. Single-cell lineage tracing has shown that GBM cells possess a high degree of plasticity. Together this suggests that phenotype switching, as opposed to genetic evolution, may be the escape mechanism that explains the failure of precision therapies to date. We profiled 86 primary-recurrent patient-matched paired GBM specimens with single-nucleus RNA, single-cell open-chromatin, DNA and spatial transcriptomic/proteomic assays. We found that recurrent GBMs are characterized by a shift to a mesenchymal phenotype. We show that the mesenchymal state is mediated by activator protein 1. Increased T-cell abundance at recurrence was prognostic and correlated with hypermutation status. We identified tumor-supportive networks of paracrine and autocrine signals between GBM cells, nonmalignant neuroglia and immune cells. We present cell-intrinsic and cell-extrinsic targets and a single-cell multiomics atlas of GBM under therapy.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Treating glioblastoma often makes a MES.Nat Cancer. 2022 Dec;3(12):1446-1448. doi: 10.1038/s43018-022-00471-1. Nat Cancer. 2022. PMID: 36539502 No abstract available.
References
-
- Mathur, R. et al. EPCO-31. Epigenomic intratumoral heterogeneity of glioblastoma in three-dimensional space. Neuro Oncol.10.1093/neuonc/noaa215.310 (2020).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
