

Canalization of genome-wide transcriptional activity in Arabidopsis thaliana accessions by MET1-dependent CG methylation
- PMID: 36539836
- PMCID: PMC9768921
- DOI: 10.1186/s13059-022-02833-5
Canalization of genome-wide transcriptional activity in Arabidopsis thaliana accessions by MET1-dependent CG methylation
Abstract
Background: Despite its conserved role on gene expression and transposable element (TE) silencing, genome-wide CG methylation differs substantially between wild Arabidopsis thaliana accessions.
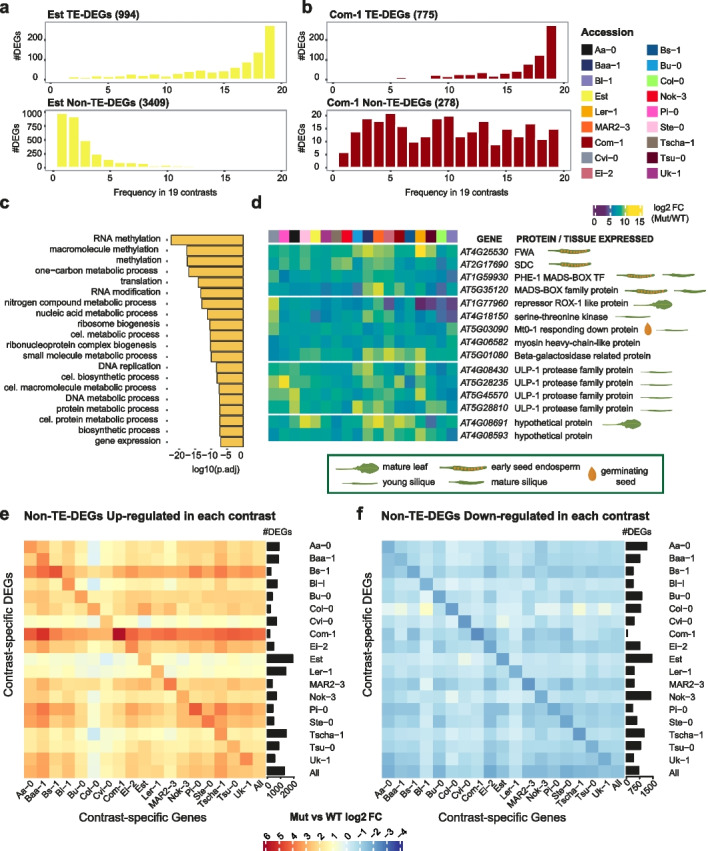
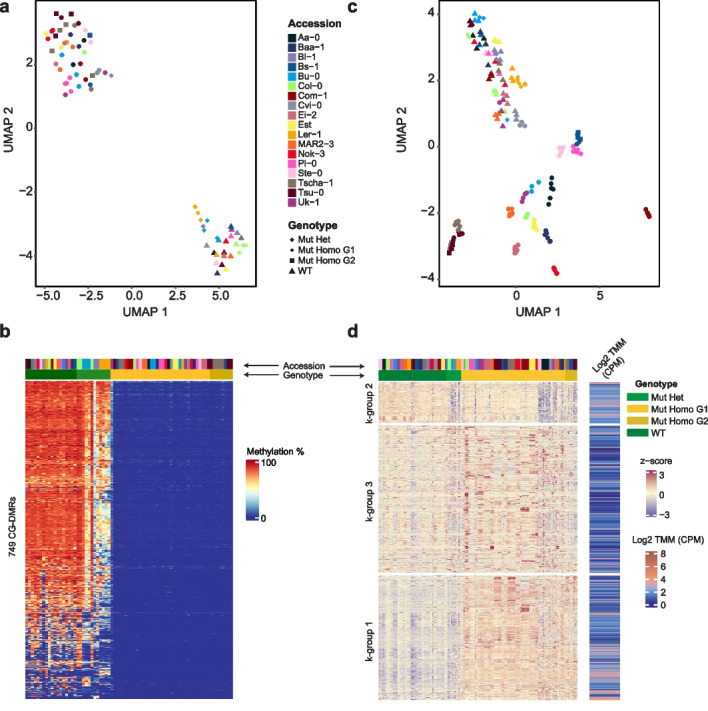
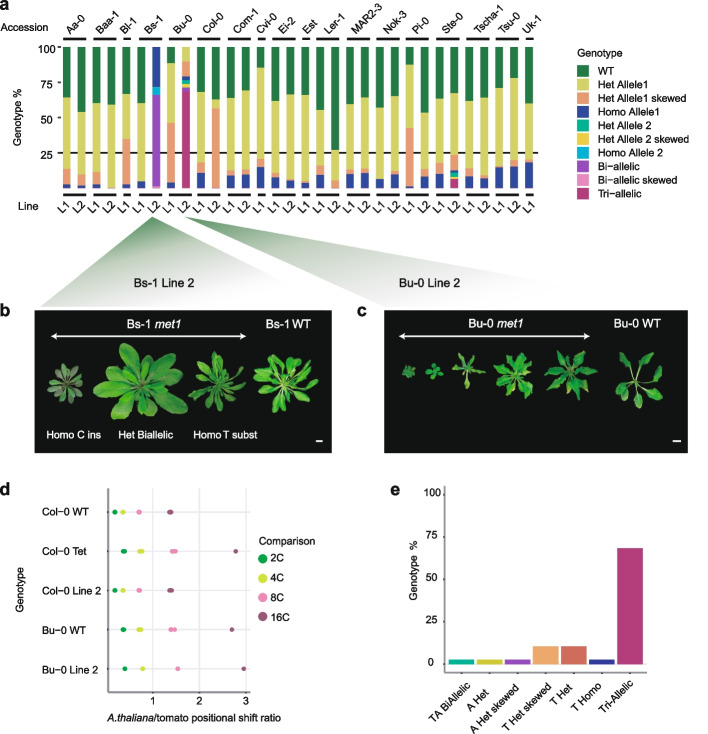
Results: To test our hypothesis that global reduction of CG methylation would reduce epigenomic, transcriptomic, and phenotypic diversity in A. thaliana accessions, we knock out MET1, which is required for CG methylation, in 18 early-flowering accessions. Homozygous met1 mutants in all accessions suffer from common developmental defects such as dwarfism and delayed flowering, in addition to accession-specific abnormalities in rosette leaf architecture, silique morphology, and fertility. Integrated analysis of genome-wide methylation, chromatin accessibility, and transcriptomes confirms that MET1 inactivation greatly reduces CG methylation and alters chromatin accessibility at thousands of loci. While the effects on TE activation are similarly drastic in all accessions, the quantitative effects on non-TE genes vary greatly. The global expression profiles of accessions become considerably more divergent from each other after genome-wide removal of CG methylation, although a few genes with diverse expression profiles across wild-type accessions tend to become more similar in mutants. Most differentially expressed genes do not exhibit altered chromatin accessibility or CG methylation in cis, suggesting that absence of MET1 can have profound indirect effects on gene expression and that these effects vary substantially between accessions.
Conclusions: Systematic analysis of MET1 requirement in different A. thaliana accessions reveals a dual role for CG methylation: for many genes, CG methylation appears to canalize expression levels, with methylation masking regulatory divergence. However, for a smaller subset of genes, CG methylation increases expression diversity beyond genetically encoded differences.
Keywords: Arabidopsis; DNA methylation; Epigenetics; Methyltransferase; Natural variation.
© 2022. The Author(s).
Conflict of interest statement
D.W. holds equity in Computomics, which advises plant breeders. D.W. consults for KWS SE, a plant breeder and seed producer. All other authors declare that they have no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
