

Predisposition to myeloid malignancies in Shwachman-Diamond syndrome: biological insights and clinical advances
- PMID: 36542827
- PMCID: PMC10082379
- DOI: 10.1182/blood.2022017739
Predisposition to myeloid malignancies in Shwachman-Diamond syndrome: biological insights and clinical advances
Abstract
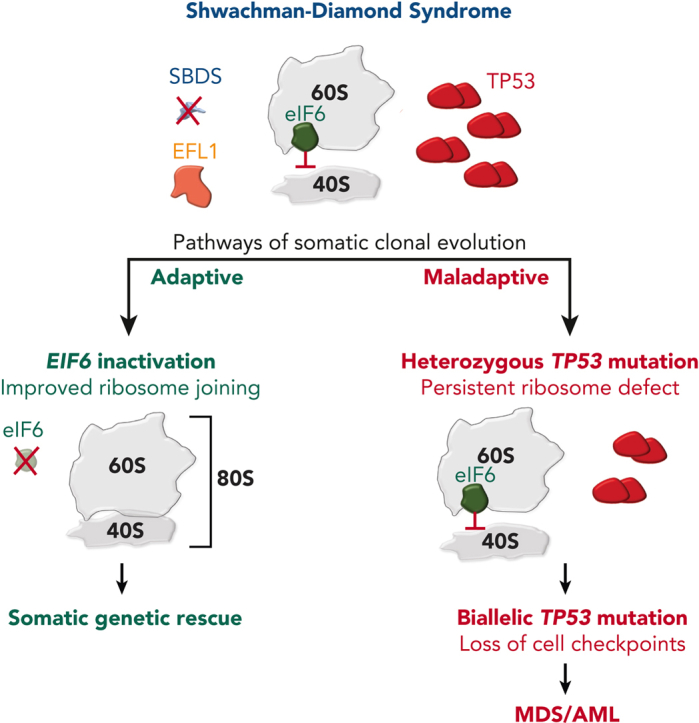
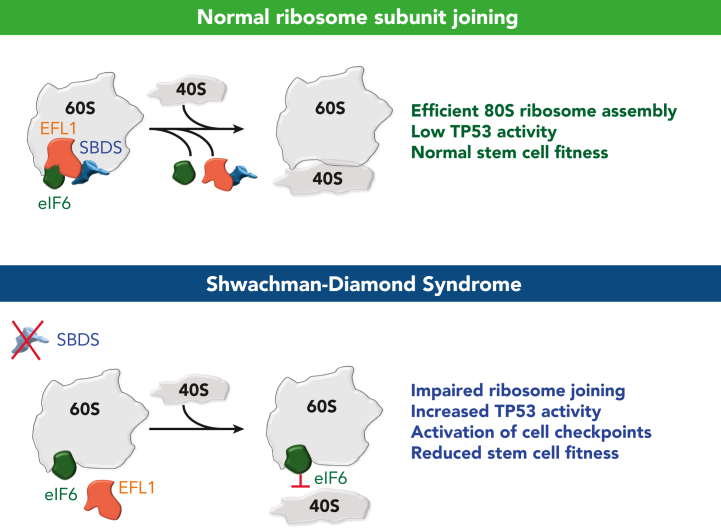
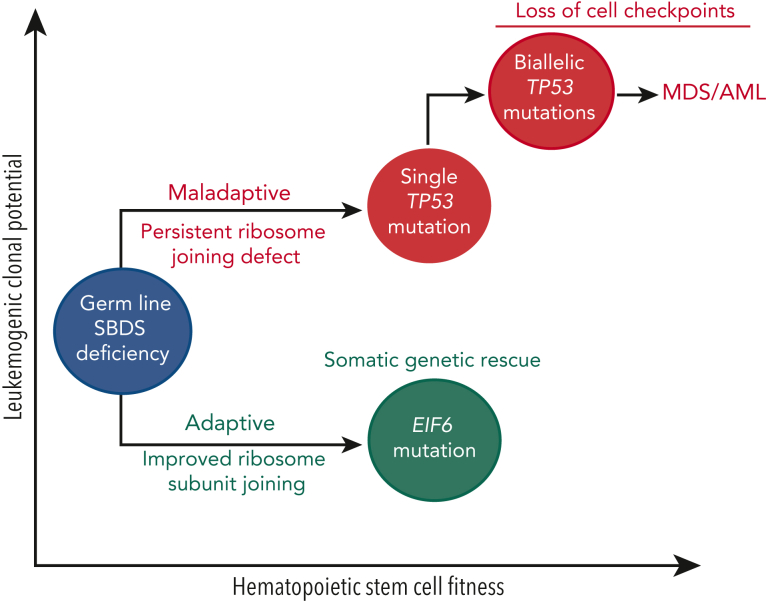
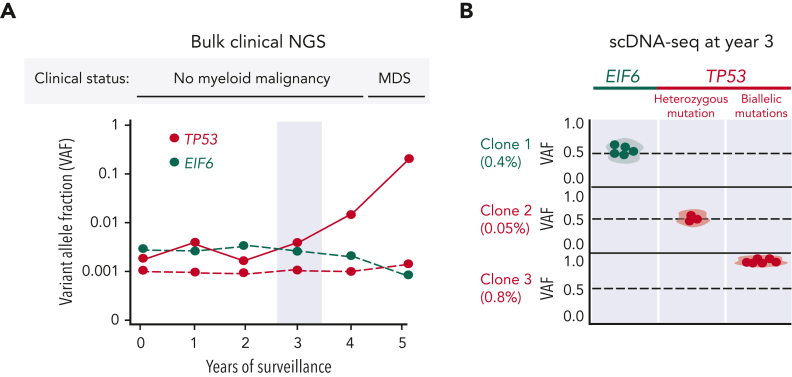
Shwachman-Diamond syndrome (SDS) is an inherited multisystem ribosomopathy characterized by exocrine pancreatic deficiency, bone marrow failure, and predisposition to myeloid malignancies. The pathobiology of SDS results from impaired ribosomal maturation due to the deficiency of SBDS and the inability to evict the antiassociation factor eIF6 from the 60S ribosomal subunit. Clinical outcomes for patients with SDS who develop myeloid malignancies are extremely poor because of high treatment-related toxicities and a high rate of refractory disease/relapse even after allogeneic hematopoietic stem cell transplant (HSCT). Registry data indicate that outcomes are improved for patients with SDS who undergo routine bone marrow surveillance and receive an HSCT before developing an overt malignancy. However, the optimal approach to hematologic surveillance and the timing of HSCT for patients with SDS is not clearly established. Recent studies have elucidated distinct patterns of somatic blood mutations in patients with SDS that either alleviate the ribosome defect via somatic rescue (heterozygous EIF6 inactivation) or disrupt cellular checkpoints, resulting in increased leukemogenic potential (heterozygous TP53 inactivation). Genomic analysis revealed that most myeloid malignancies in patients with SDS have biallelic loss-of-function TP53 mutations. Single-cell DNA sequencing of SDS bone marrow samples can detect premalignant biallelic TP53-mutated clones before clinical diagnosis, suggesting that molecular surveillance may enhance the detection of incipient myeloid malignancies when HSCT may be most effective. Here, we review the clinical, genetic, and biologic features of SDS. In addition, we present evidence supporting the hematologic surveillance for patients with SDS that incorporates clinical, pathologic, and molecular data to risk stratify patients and prioritize transplant evaluation for patients with SDS with high-risk features.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: C.R.R. has provided consulting for RenBio. A.S. declares no competing financial interests.
Figures
Comment in
-
Introduction to a review series on germ line predisposition to hematologic malignancies: time to consider germ line testing.Blood. 2023 Mar 30;141(13):1509-1512. doi: 10.1182/blood.2023019846. Blood. 2023. PMID: 36787501 No abstract available.
References
-
- Nelson AS, Myers KC. Diagnosis, treatment, and molecular pathology of Shwachman-Diamond syndrome. Hematol Oncol Clin North Am. 2018;32(4):687–700. - PubMed
-
- Boocock GRB, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman–Diamond syndrome. Nat Genet. 2002;33(1):97–101. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
