

Differential diagnosis of bone marrow failure syndromes guided by machine learning
- PMID: 36542832
- PMCID: PMC10163315
- DOI: 10.1182/blood.2022017518
Differential diagnosis of bone marrow failure syndromes guided by machine learning
Abstract
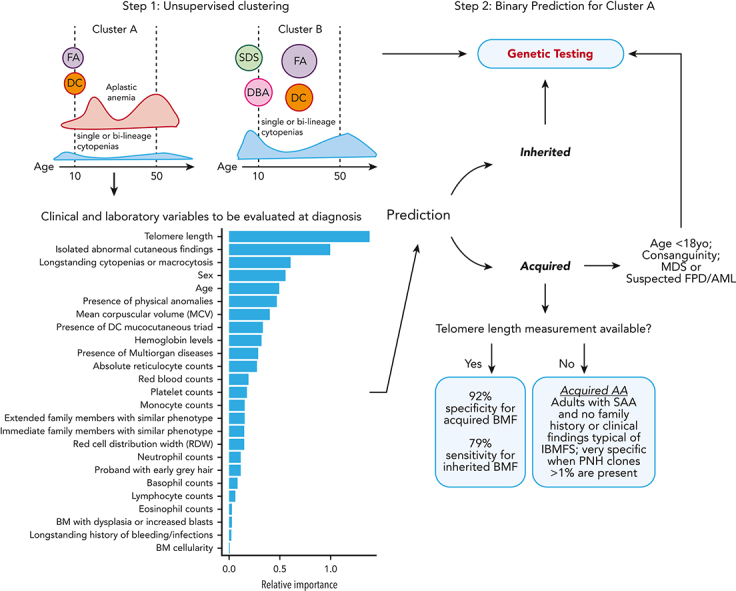
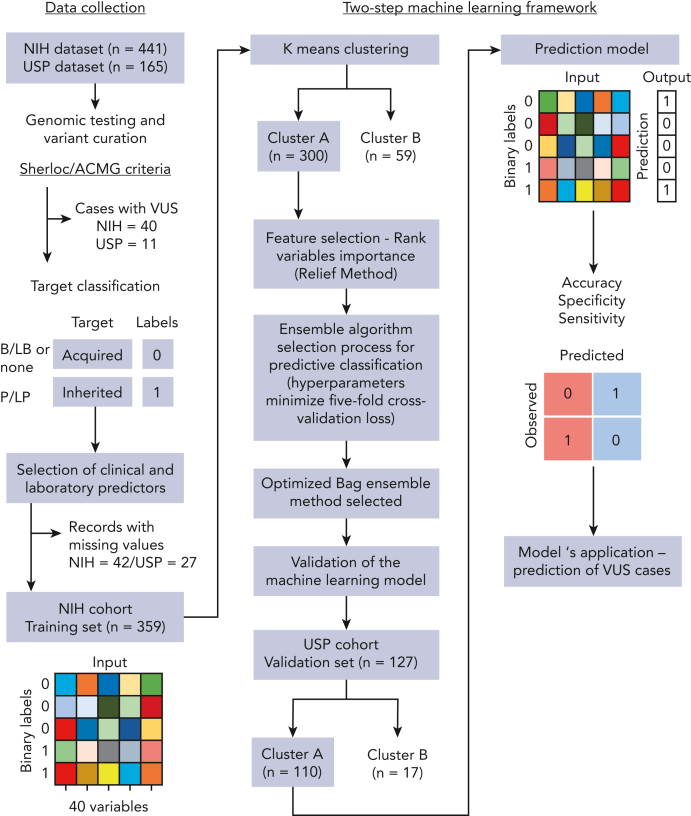
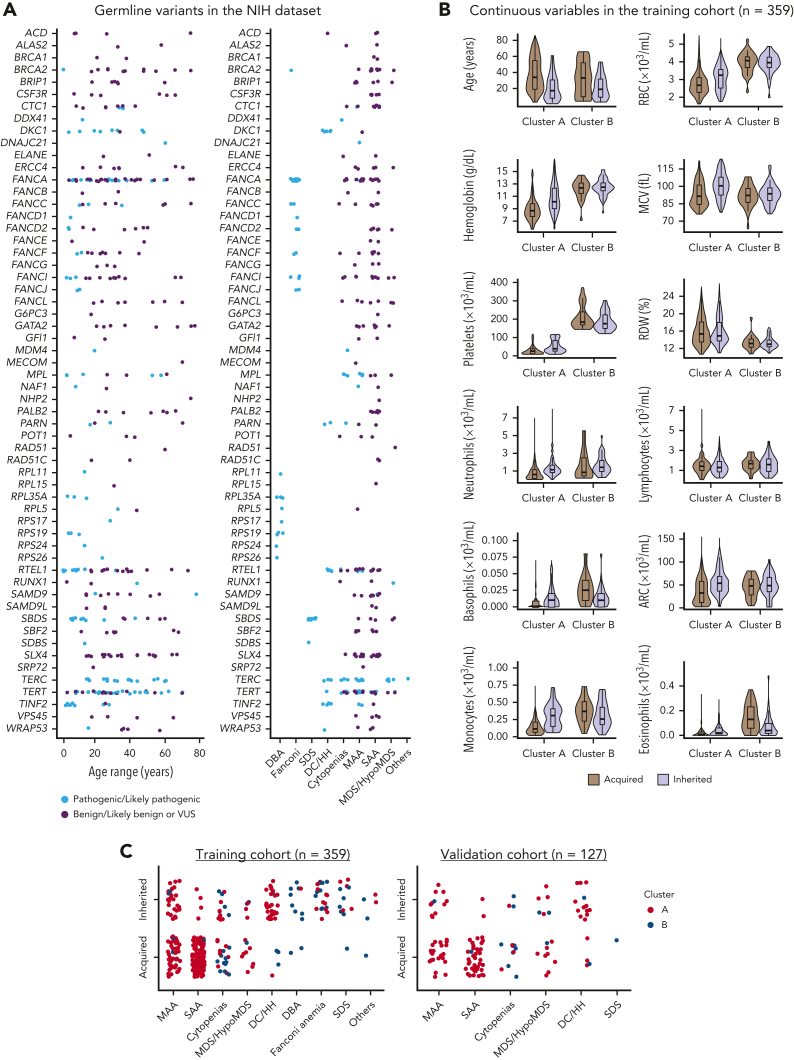
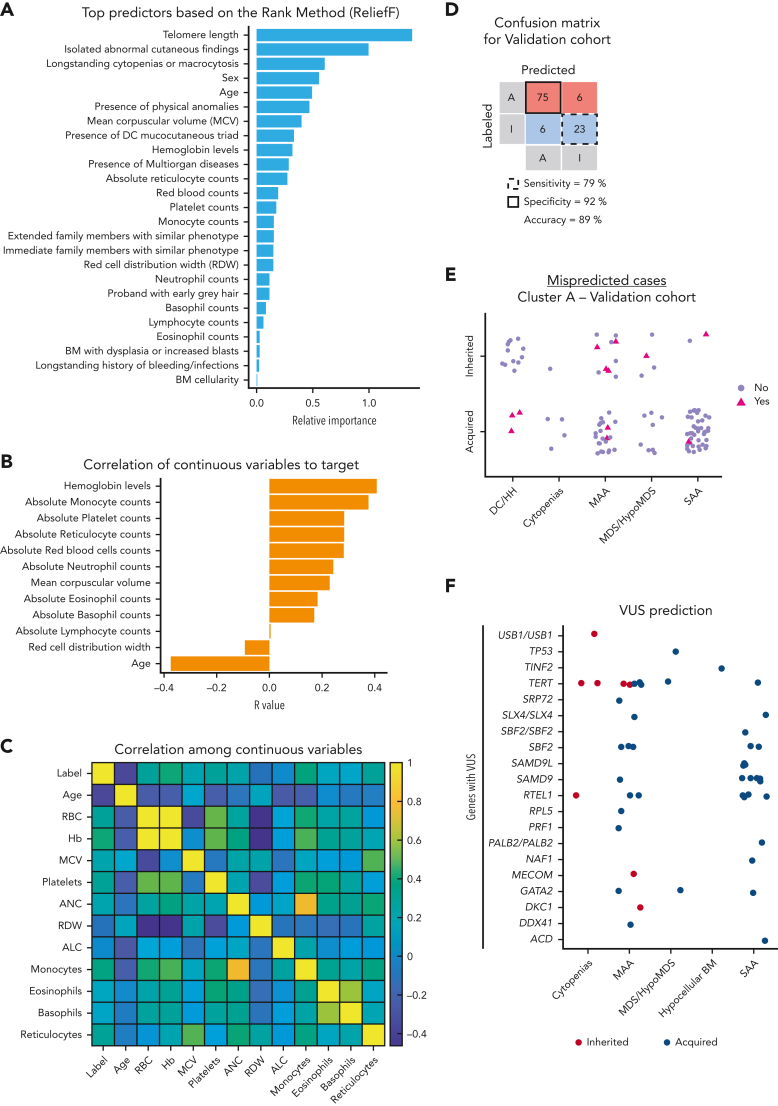
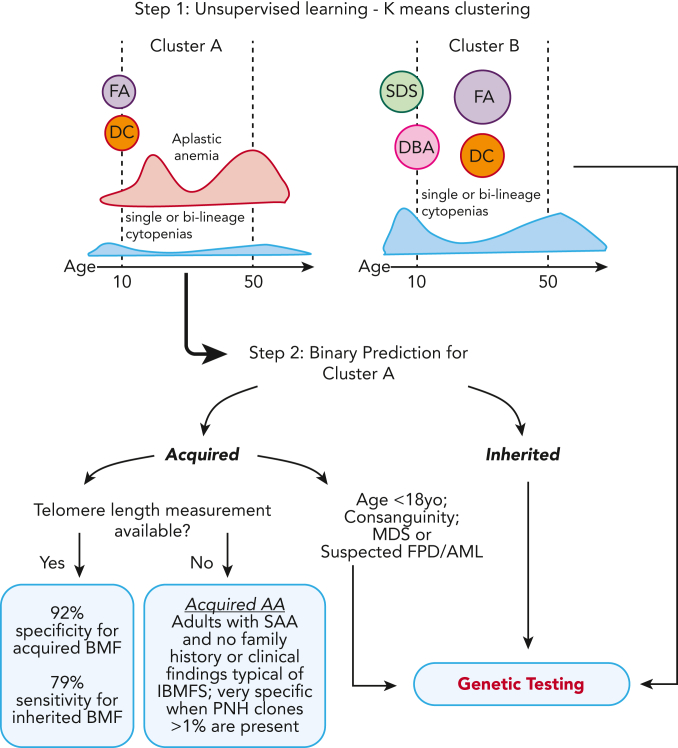
The choice to postpone treatment while awaiting genetic testing can result in significant delay in definitive therapies in patients with severe pancytopenia. Conversely, the misdiagnosis of inherited bone marrow failure (BMF) can expose patients to ineffectual and expensive therapies, toxic transplant conditioning regimens, and inappropriate use of an affected family member as a stem cell donor. To predict the likelihood of patients having acquired or inherited BMF, we developed a 2-step data-driven machine-learning model using 25 clinical and laboratory variables typically recorded at the initial clinical encounter. For model development, patients were labeled as having acquired or inherited BMF depending on their genomic data. Data sets were unbiasedly clustered, and an ensemble model was trained with cases from the largest cluster of a training cohort (n = 359) and validated with an independent cohort (n = 127). Cluster A, the largest group, was mostly immune or inherited aplastic anemia, whereas cluster B comprised underrepresented BMF phenotypes and was not included in the next step of data modeling because of a small sample size. The ensemble cluster A-specific model was accurate (89%) to predict BMF etiology, correctly predicting inherited and likely immune BMF in 79% and 92% of cases, respectively. Our model represents a practical guide for BMF diagnosis and highlights the importance of clinical and laboratory variables in the initial evaluation, particularly telomere length. Our tool can be potentially used by general hematologists and health care providers not specialized in BMF, and in under-resourced centers, to prioritize patients for genetic testing or for expeditious treatment.
Conflict of interest statement
Conflict-of-interest disclosure: N.S.Y. received research funding from Novartis by way of a Cooperative Research and Development Agreement. R.M.S. received royalties from cad, Ping An, Philips, Scan Med, and Translation Holdings and his laboratory received research support from Ping An and NVIDIA. Y.T. is currently employed by Ping An. The remaining authors declare no competing financial interests.
Figures
Comment in
-
AI for AA: machine learning makes an entry.Blood. 2023 Apr 27;141(17):2040-2042. doi: 10.1182/blood.2022019444. Blood. 2023. PMID: 37103950 No abstract available.
References
-
- Bluteau O, Sebert M, Leblanc T, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. 2018;131(7):717–732. - PubMed
-
- Wegman-Ostrosky T, Savage SA. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol. 2017;177(4):526–542. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
