

Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation
- PMID: 36543887
- PMCID: PMC9768794
- DOI: 10.1038/s41573-022-00612-2
Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation
Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating disease caused by degeneration of motor neurons. As with all major neurodegenerative disorders, development of disease-modifying therapies has proven challenging for multiple reasons. Nevertheless, ALS is one of the few neurodegenerative diseases for which disease-modifying therapies are approved. Significant discoveries and advances have been made in ALS preclinical models, genetics, pathology, biomarkers, imaging and clinical readouts over the last 10-15 years. At the same time, novel therapeutic paradigms are being applied in areas of high unmet medical need, including neurodegenerative disorders. These developments have evolved our knowledge base, allowing identification of targeted candidate therapies for ALS with diverse mechanisms of action. In this Review, we discuss how this advanced knowledge, aligned with new approaches, can enable effective translation of therapeutic agents from preclinical studies through to clinical benefit for patients with ALS. We anticipate that this approach in ALS will also positively impact the field of drug discovery for neurodegenerative disorders more broadly.
© 2022. Springer Nature Limited.
Conflict of interest statement
R.J.M. is cofounder of and holds shares in Keapstone Therapeutics, collaborates and receives funding from BenevolentAI, Quell Therapeutics, Sosei Heptares and MSD, is a consultant to Aclipse Therapeutics, has shares in Aclipse One Inc and is an inventor on patents related to M102. N.S. is an employee and shareholder of Aclipse Therapeutics. H.J.R. is the chairman of the Board of Aclipse Therapeutics. F.M. is an employee and shareholder of Merck and Co. P.J.S. is an advisory board member and consultant for Biogen, Aclipse Therapeutics, Quell Therapeutics, BenevolentAI, QurAlis, Astex, GeniUS and Eli Lilly and collaborates with and receives research funding from Quell Therapeutics, Aclipse Therapeutics, Pfizer and SwanBio. She is a cofounder of and holds shares in Keapstone Therapeutics and holds shares in Aclipse One Inc. She is an inventor on patents related to low-dose IL-2, SRSF1 and M102. Support for clinical trials participation in the last five years has been received from Biogen, Alexion, Orion Pharma, WAVE, the EU Horizon 2020 programme and UK NIHR.
Figures
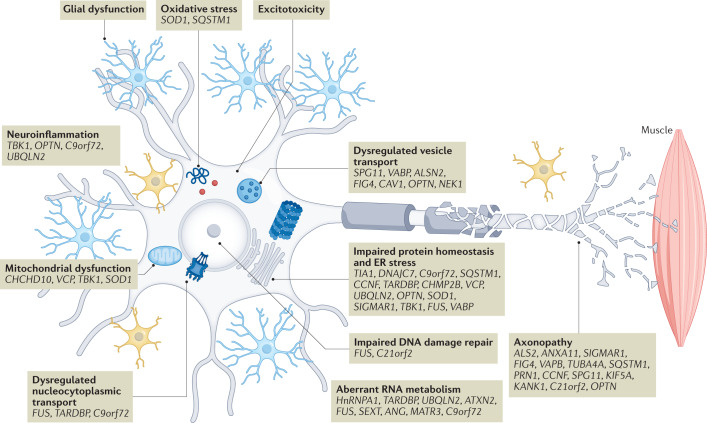

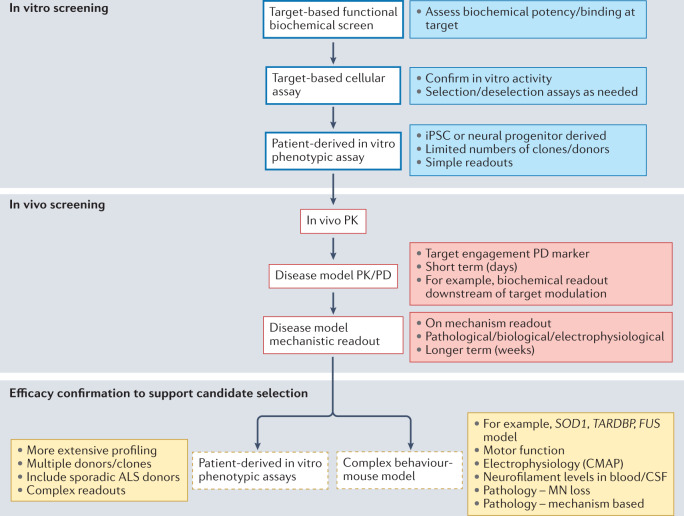
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
