

An altered extracellular matrix-integrin interface contributes to Huntington's disease-associated CNS dysfunction in glial and vascular cells
- PMID: 36547263
- PMCID: PMC10117161
- DOI: 10.1093/hmg/ddac303
An altered extracellular matrix-integrin interface contributes to Huntington's disease-associated CNS dysfunction in glial and vascular cells
Abstract
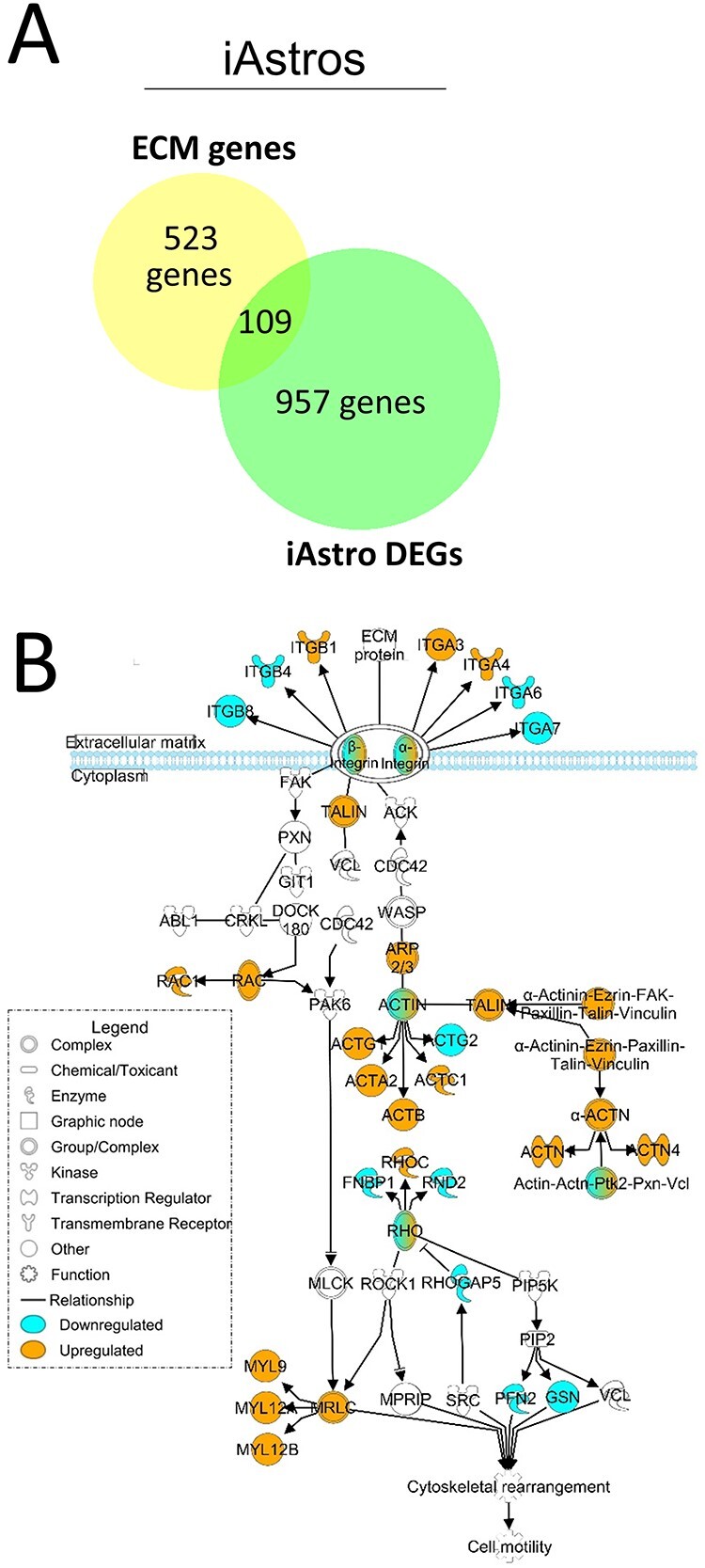
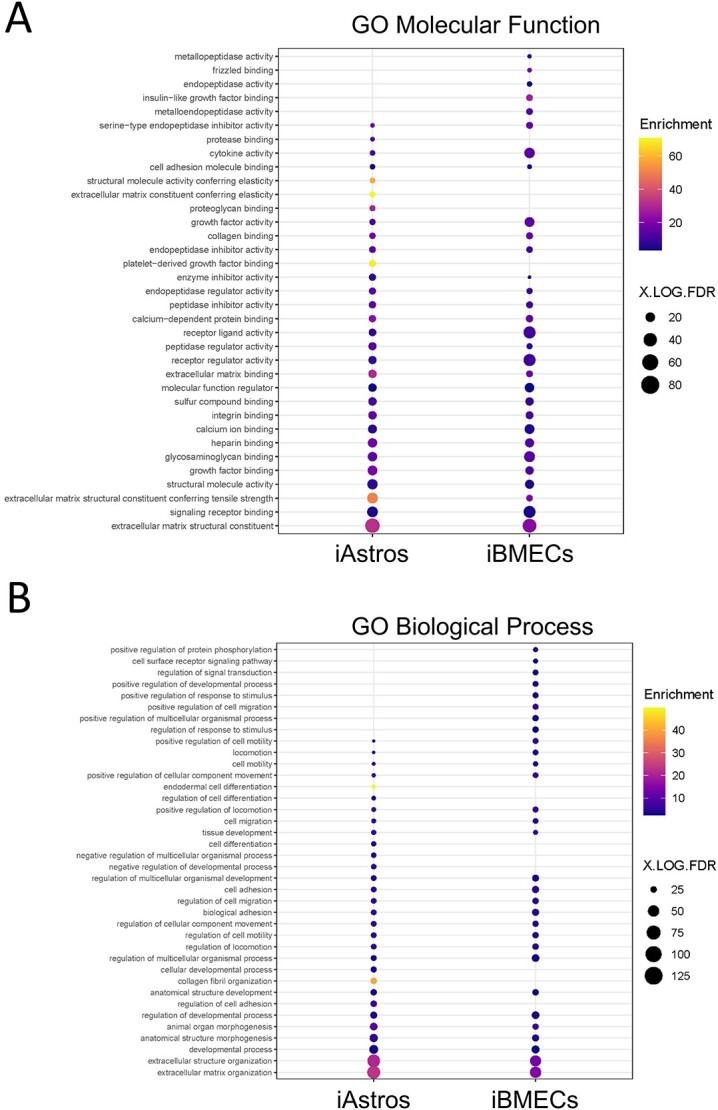
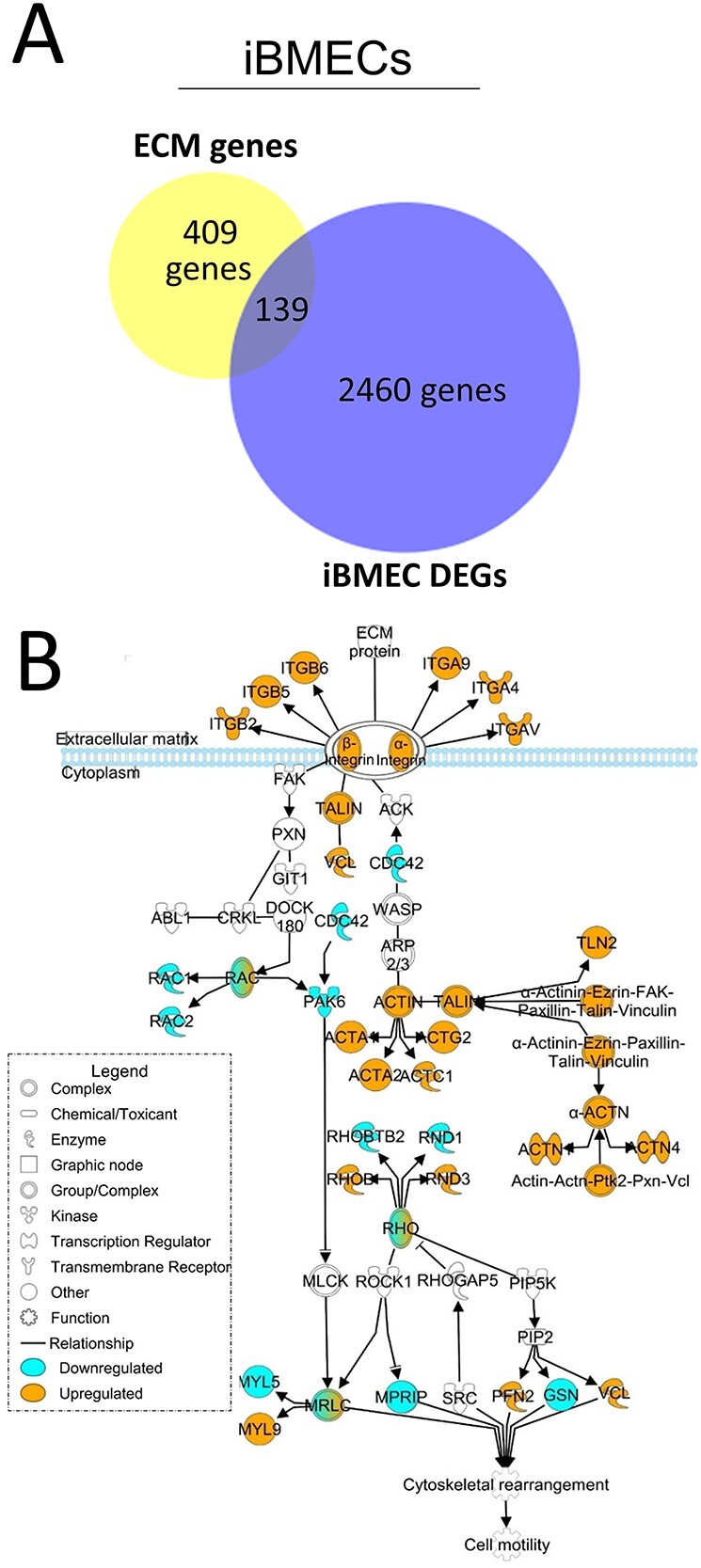
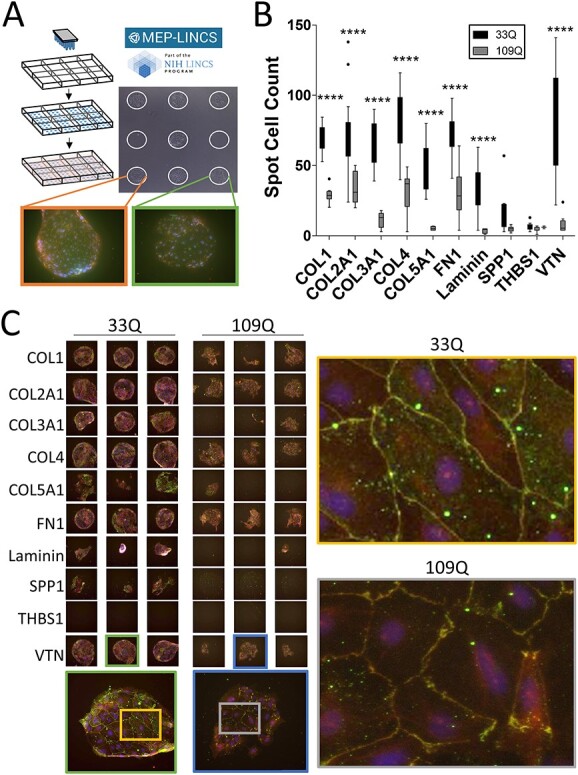
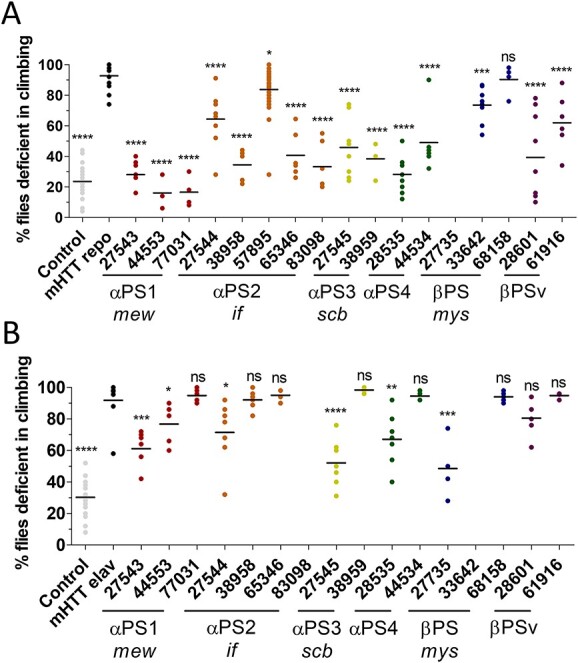
Astrocytes and brain endothelial cells are components of the neurovascular unit that comprises the blood-brain barrier (BBB) and their dysfunction contributes to pathogenesis in Huntington's disease (HD). Defining the contribution of these cells to disease can inform cell-type-specific effects and uncover new disease-modifying therapeutic targets. These cells express integrin (ITG) adhesion receptors that anchor the cells to the extracellular matrix (ECM) to maintain the integrity of the BBB. We used HD patient-derived induced pluripotent stem cell (iPSC) modeling to study the ECM-ITG interface in astrocytes and brain microvascular endothelial cells and found ECM-ITG dysregulation in human iPSC-derived cells that may contribute to the dysfunction of the BBB in HD. This disruption has functional consequences since reducing ITG expression in glia in an HD Drosophila model suppressed disease-associated CNS dysfunction. Since ITGs can be targeted therapeutically and manipulating ITG signaling prevents neurodegeneration in other diseases, defining the role of ITGs in HD may provide a novel strategy of intervention to slow CNS pathophysiology to treat HD.
© The Author(s) 2022. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures
References
-
- MacDonald, M.E., Ambrose, C.M., Duyao, M.P., Myers, R.H., Lin, C., Srinidhi, L., Barnes, G., Taylor, S.A., James, M. and Groot, N. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell, 72, 971–983. - PubMed
-
- Ross, C.A. and Tabrizi, S.J. (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol., 10, 83–98. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
