

Cross-kingdom synthetic microbiota supports tomato suppression of Fusarium wilt disease
- PMID: 36550095
- PMCID: PMC9780251
- DOI: 10.1038/s41467-022-35452-6
Cross-kingdom synthetic microbiota supports tomato suppression of Fusarium wilt disease
Erratum in
-
Author Correction: Cross-kingdom synthetic microbiota supports tomato suppression of Fusarium wilt disease.Nat Commun. 2023 Oct 24;14(1):6755. doi: 10.1038/s41467-023-42557-z. Nat Commun. 2023. PMID: 37875479 Free PMC article. No abstract available.
Abstract
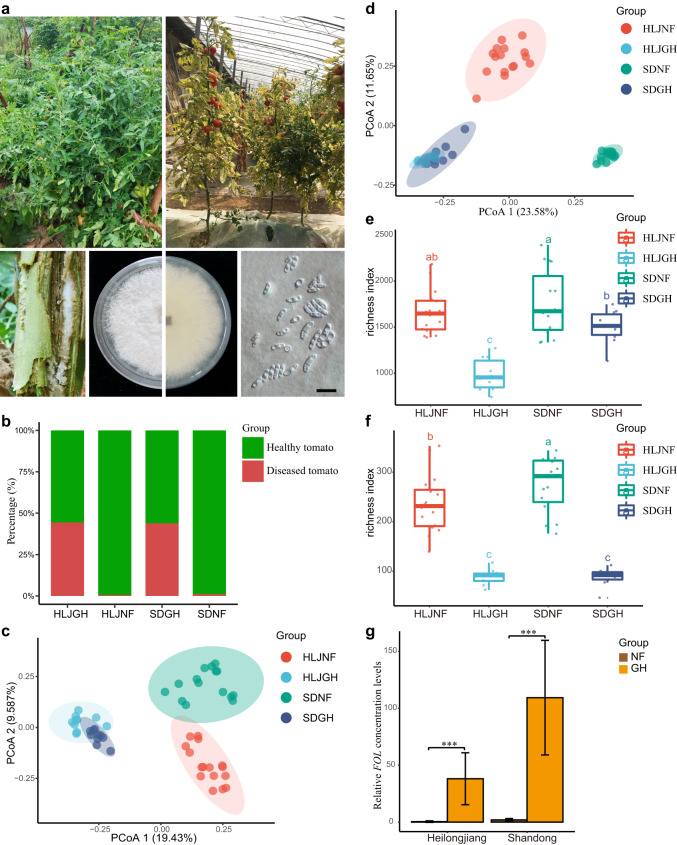
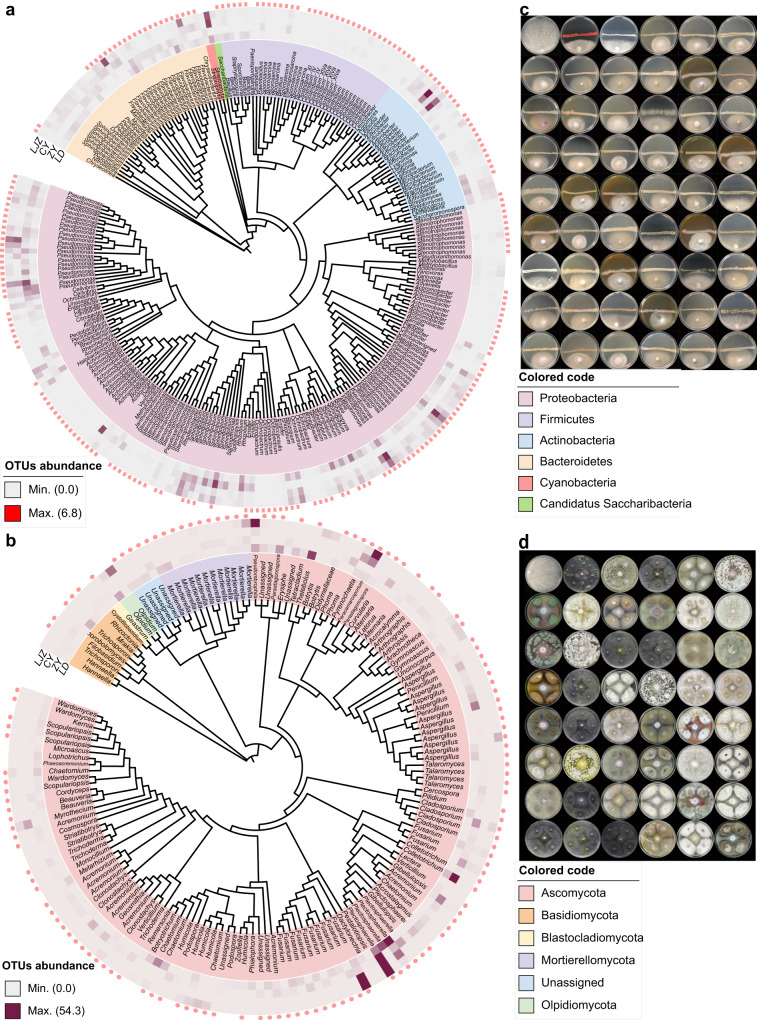
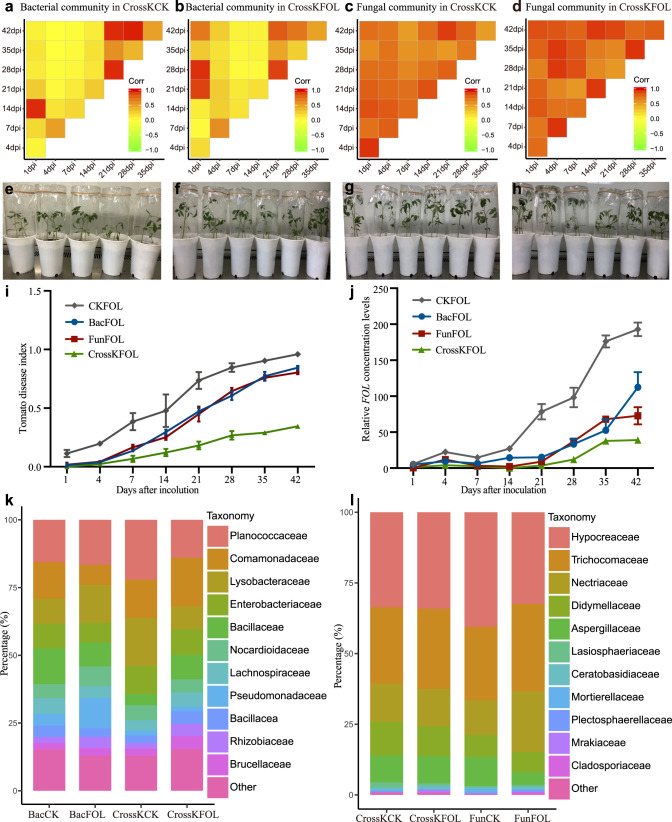
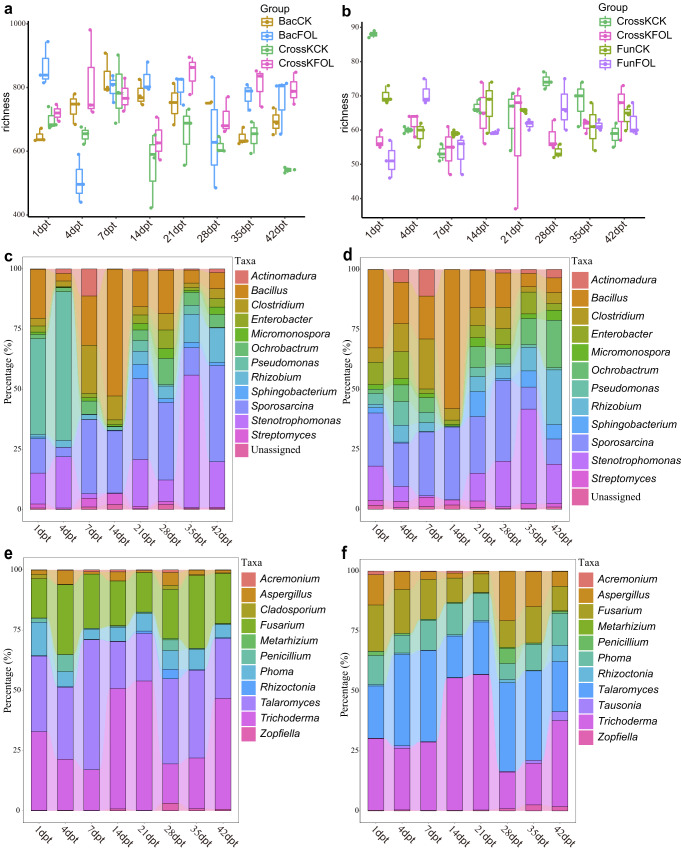
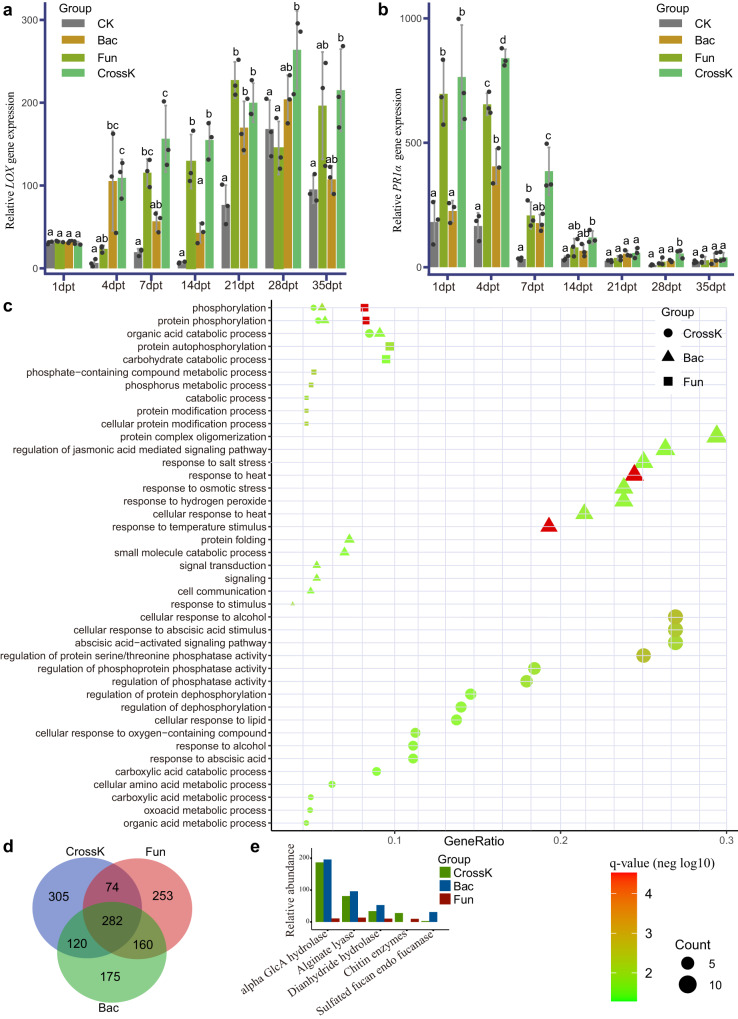
The role of rhizosphere microbiota in the resistance of tomato plant against soil-borne Fusarium wilt disease (FWD) remains unclear. Here, we showed that the FWD incidence was significantly negatively correlated with the diversity of both rhizosphere bacterial and fungal communities. Using the microbiological culturomic approach, we selected 205 unique strains to construct different synthetic communities (SynComs), which were inoculated into germ-free tomato seedlings, and their roles in suppressing FWD were monitored using omics approach. Cross-kingdom (fungi and bacteria) SynComs were most effective in suppressing FWD than those of Fungal or Bacterial SynComs alone. This effect was underpinned by a combination of molecular mechanisms related to plant immunity and microbial interactions contributed by the bacterial and fungal communities. This study provides new insight into the dynamics of microbiota in pathogen suppression and host immunity interactions. Also, the formulation and manipulation of SynComs for functional complementation constitute a beneficial strategy in controlling soil-borne disease.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Rhizosphere-enriched microbes as a pool to design synthetic communities for reproducible beneficial outputs.FEMS Microbiol Ecol. 2019 Oct 1;95(10):fiz138. doi: 10.1093/femsec/fiz138. FEMS Microbiol Ecol. 2019. PMID: 31504462
-
Deciphering the Synergies of Reductive Soil Disinfestation Combined with Biochar and Antagonistic Microbial Inoculation in Cucumber Fusarium Wilt Suppression Through Rhizosphere Microbiota Structure.Microb Ecol. 2023 Apr;85(3):980-997. doi: 10.1007/s00248-022-02097-3. Epub 2022 Aug 11. Microb Ecol. 2023. PMID: 35948832
-
The rhizosphere microbial community in a multiple parallel mineralization system suppresses the pathogenic fungus Fusarium oxysporum.Microbiologyopen. 2013 Dec;2(6):997-1009. doi: 10.1002/mbo3.140. Epub 2013 Nov 8. Microbiologyopen. 2013. PMID: 24311557 Free PMC article.
-
Advances in Rhizosphere Microbiome and Rhizosphere Immunity Effect: A Review.J Agric Food Chem. 2025 Jun 18;73(24):14707-14721. doi: 10.1021/acs.jafc.5c02003. Epub 2025 Jun 3. J Agric Food Chem. 2025. PMID: 40459209 Review.
-
Microbiome-Mediated Strategies to Manage Major Soil-Borne Diseases of Tomato.Plants (Basel). 2024 Jan 25;13(3):364. doi: 10.3390/plants13030364. Plants (Basel). 2024. PMID: 38337897 Free PMC article. Review.
Cited by
-
Root hair developmental regulators orchestrate drought triggered microbiome changes and the interaction with beneficial Rhizobiaceae.Nat Commun. 2024 Nov 20;15(1):10068. doi: 10.1038/s41467-024-54417-5. Nat Commun. 2024. PMID: 39567534 Free PMC article.
-
Differential analysis of the quality and soil microhabitat of Epimedium koreanum Nakai under different cultivation methods.Front Microbiol. 2025 May 22;16:1556173. doi: 10.3389/fmicb.2025.1556173. eCollection 2025. Front Microbiol. 2025. PMID: 40475383 Free PMC article.
-
Community standards and future opportunities for synthetic communities in plant-microbiota research.Nat Microbiol. 2024 Nov;9(11):2774-2784. doi: 10.1038/s41564-024-01833-4. Epub 2024 Oct 30. Nat Microbiol. 2024. PMID: 39478084 Review.
-
The Cultured Microbiome of Pollinated Maize Silks Shifts after Infection with Fusarium graminearum and Varies by Distance from the Site of Pathogen Inoculation.Pathogens. 2023 Nov 6;12(11):1322. doi: 10.3390/pathogens12111322. Pathogens. 2023. PMID: 38003787 Free PMC article.
-
Bacillus velezensis LT1: a potential biocontrol agent for southern blight on Coptis chinensis.Front Microbiol. 2024 Mar 4;15:1337655. doi: 10.3389/fmicb.2024.1337655. eCollection 2024. Front Microbiol. 2024. PMID: 38500587 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
