

Efficient combinatorial targeting of RNA transcripts in single cells with Cas13 RNA Perturb-seq
- PMID: 36550277
- PMCID: PMC10030154
- DOI: 10.1038/s41592-022-01705-x
Efficient combinatorial targeting of RNA transcripts in single cells with Cas13 RNA Perturb-seq
Abstract
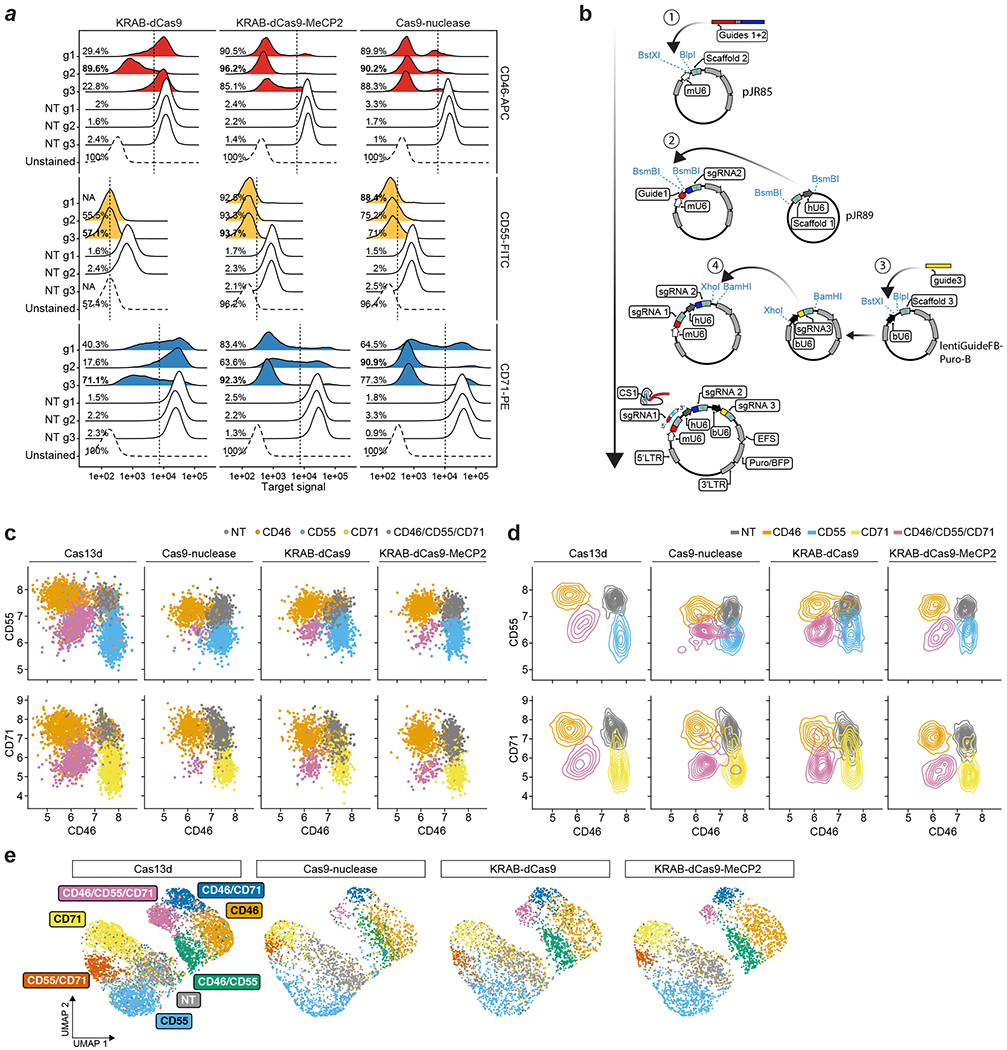
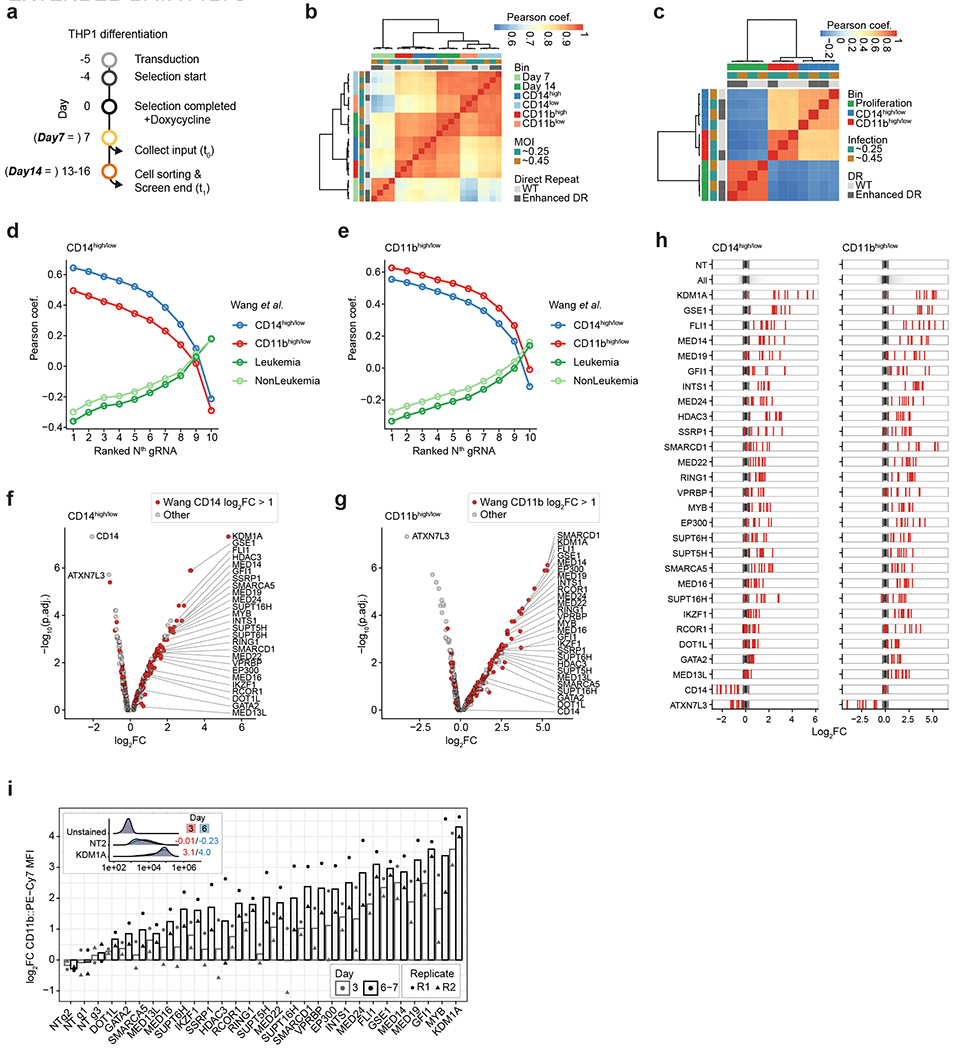
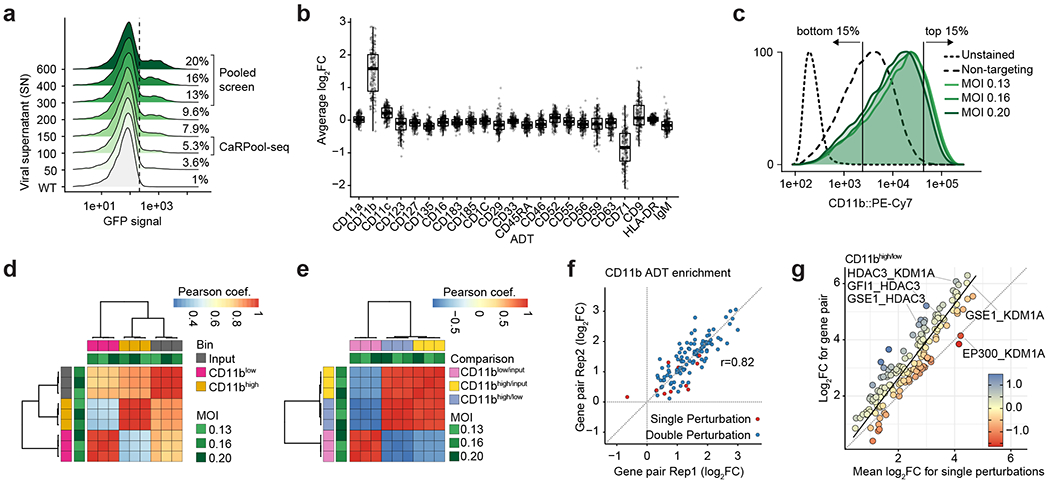
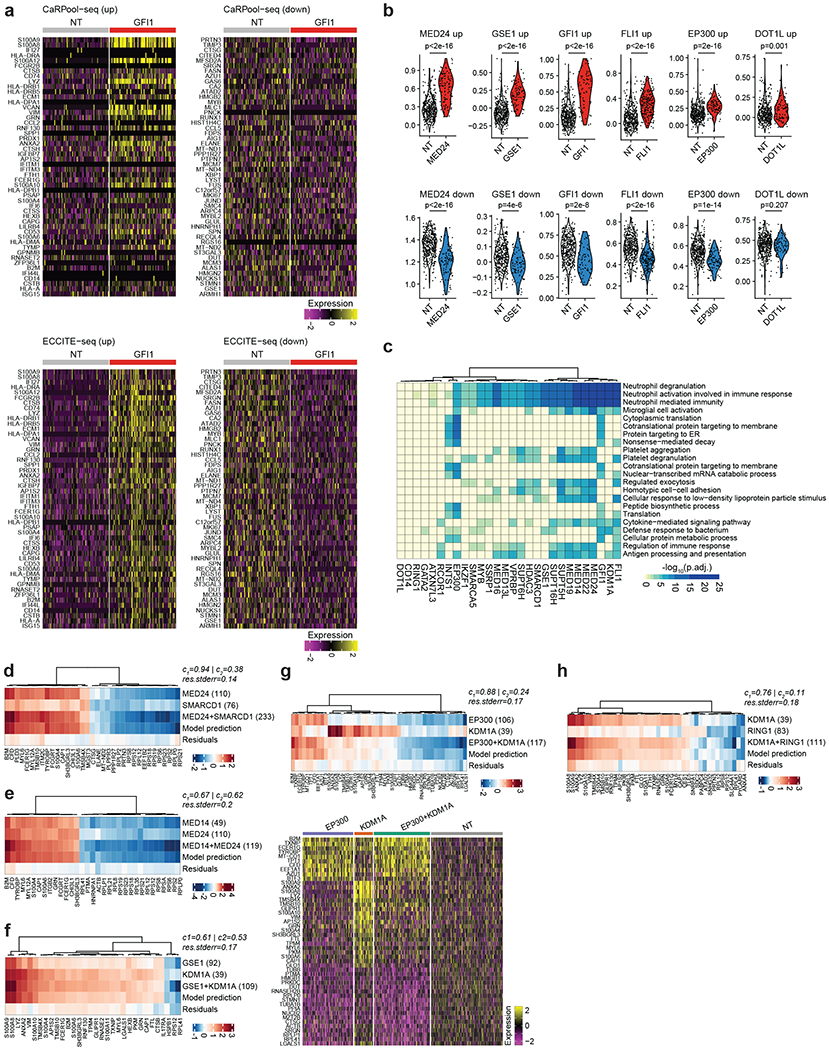
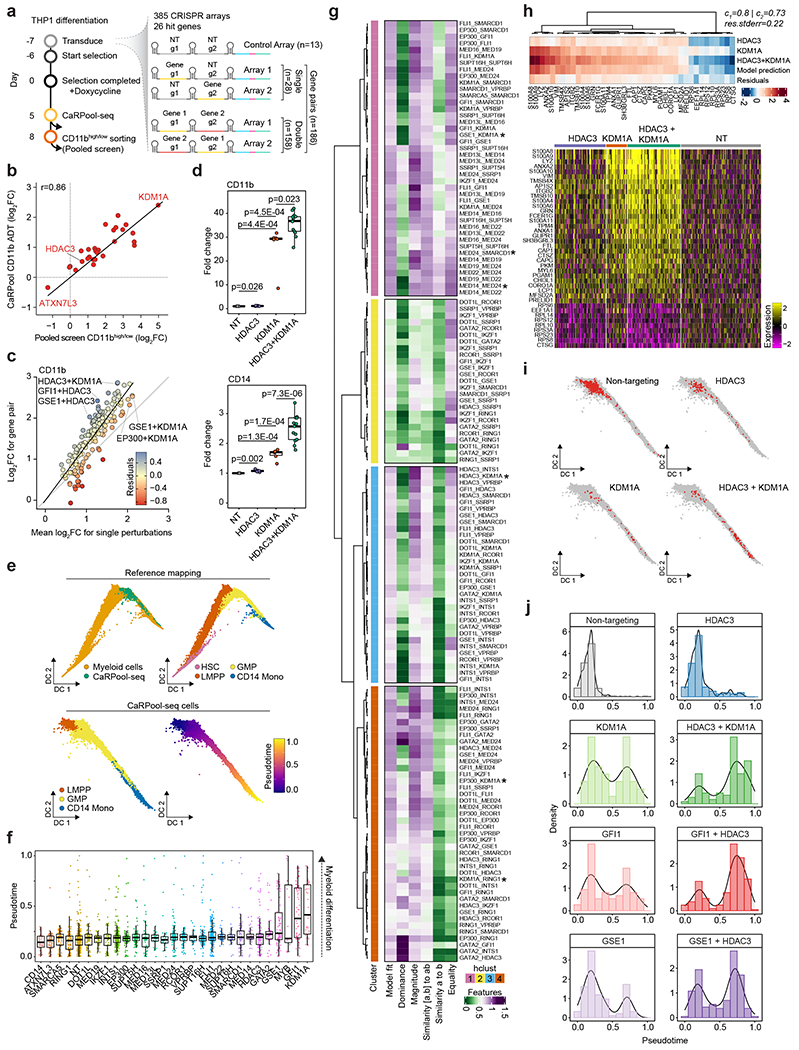
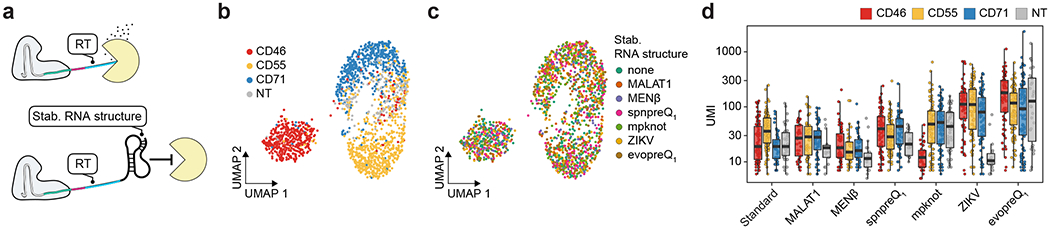
Pooled CRISPR screens coupled with single-cell RNA-sequencing have enabled systematic interrogation of gene function and regulatory networks. Here, we introduce Cas13 RNA Perturb-seq (CaRPool-seq), which leverages the RNA-targeting CRISPR-Cas13d system and enables efficient combinatorial perturbations alongside multimodal single-cell profiling. CaRPool-seq encodes multiple perturbations on a cleavable CRISPR array that is associated with a detectable barcode sequence, allowing for the simultaneous targeting of multiple genes. We compared CaRPool-seq to existing Cas9-based methods, highlighting its unique strength to efficiently profile combinatorially perturbed cells. Finally, we apply CaRPool-seq to perform multiplexed combinatorial perturbations of myeloid differentiation regulators in an acute myeloid leukemia (AML) model system and identify extensive interactions between different chromatin regulators that can enhance or suppress AML differentiation phenotypes.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
In the past 3 years, R.S. has worked as a consultant for Bristol-Myers Squibb, Regeneron and Kallyope, and served as a SAB member for ImmunAI, Apollo Life Sciences GmbH, Nanostring and the New York City Pandemic Response Laboratory. N.E.S. is an advisor to Vertex and QIAGEN and is a cofounder of OverT Bio. P.S. is a coinventor on a patent related to protein detection by sequencing as described in this work. The New York Genome Center and New York University have applied for patents relating to the work in this article. The remaining authors declare no competing interests.
Figures
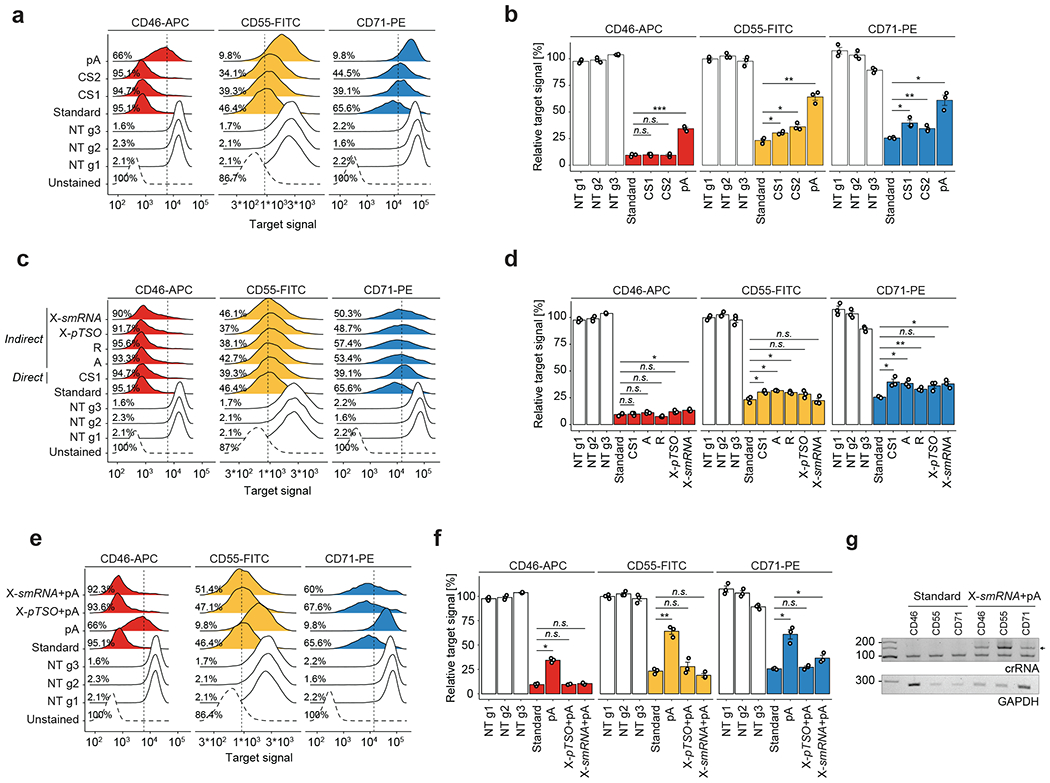
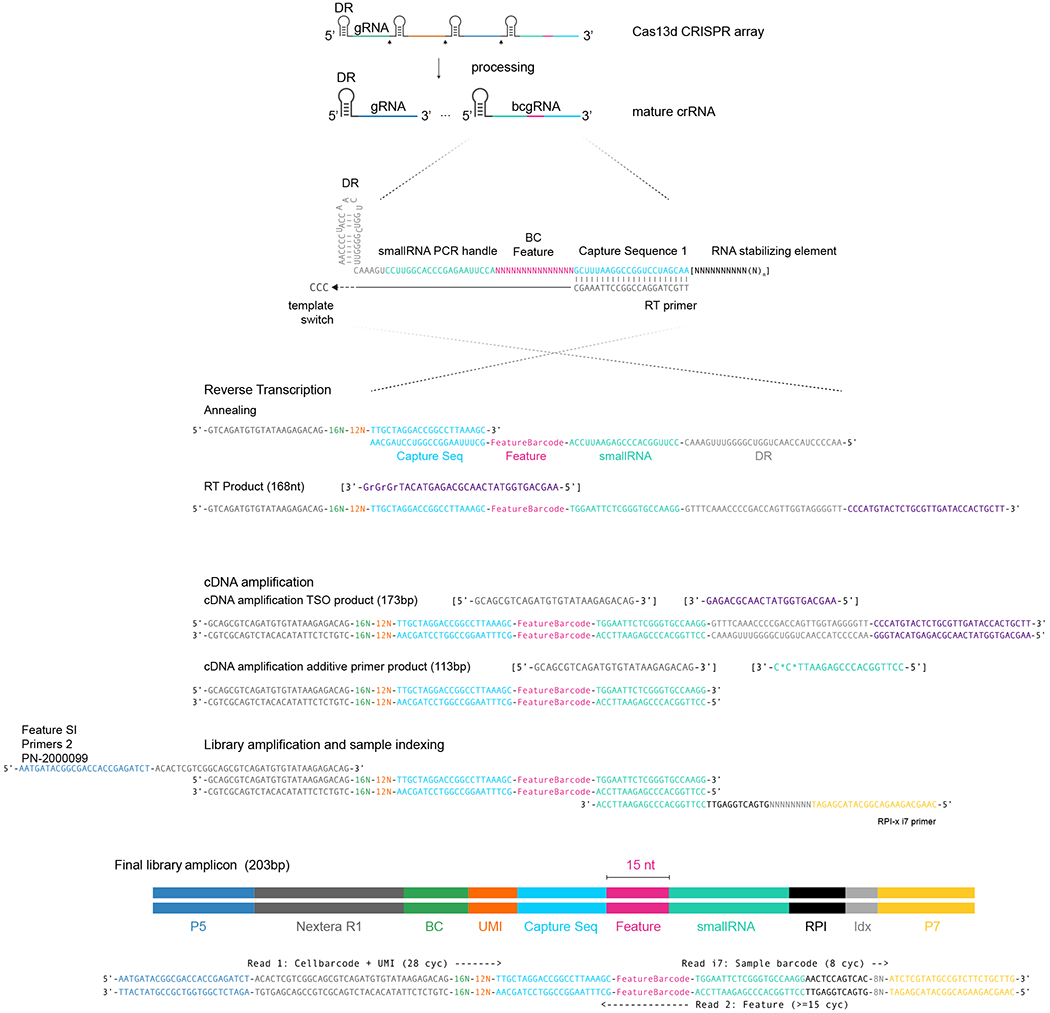
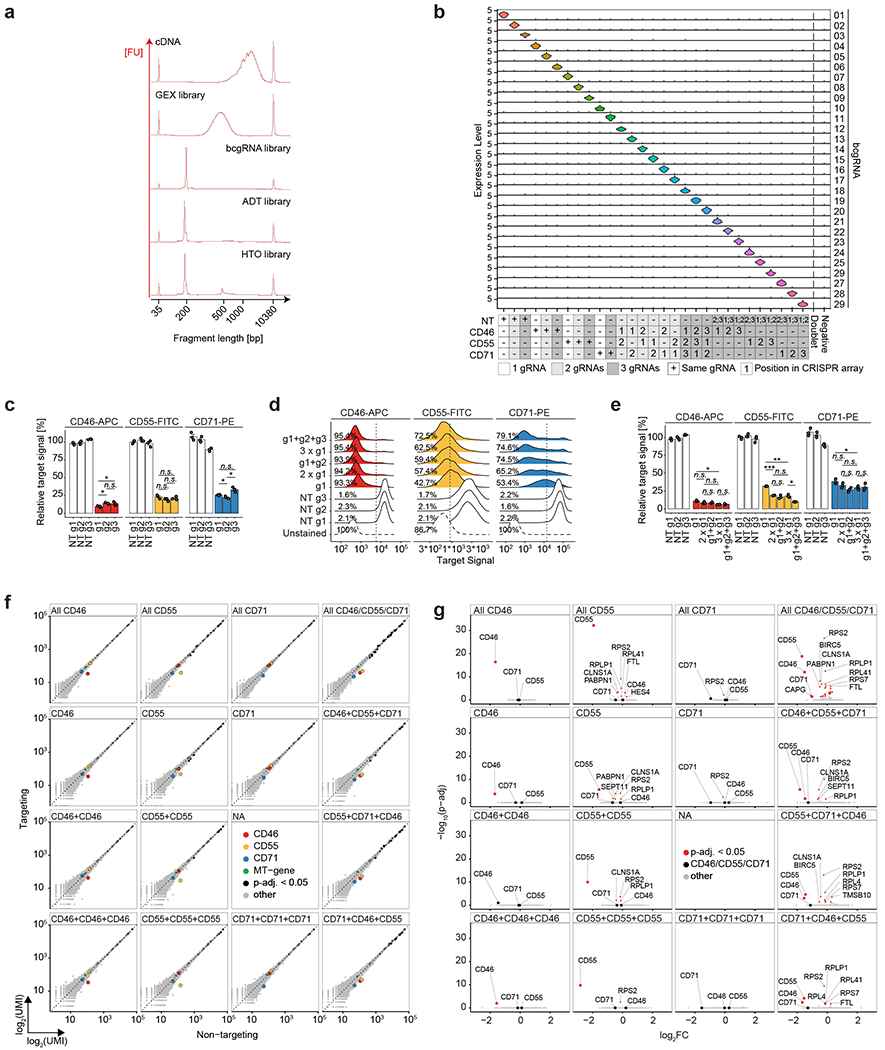
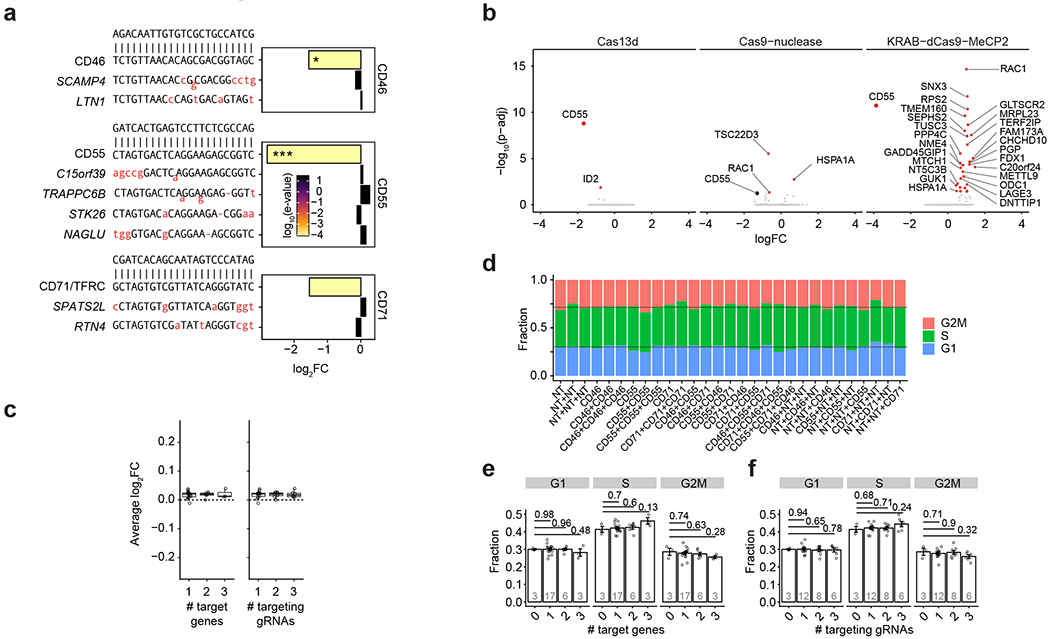
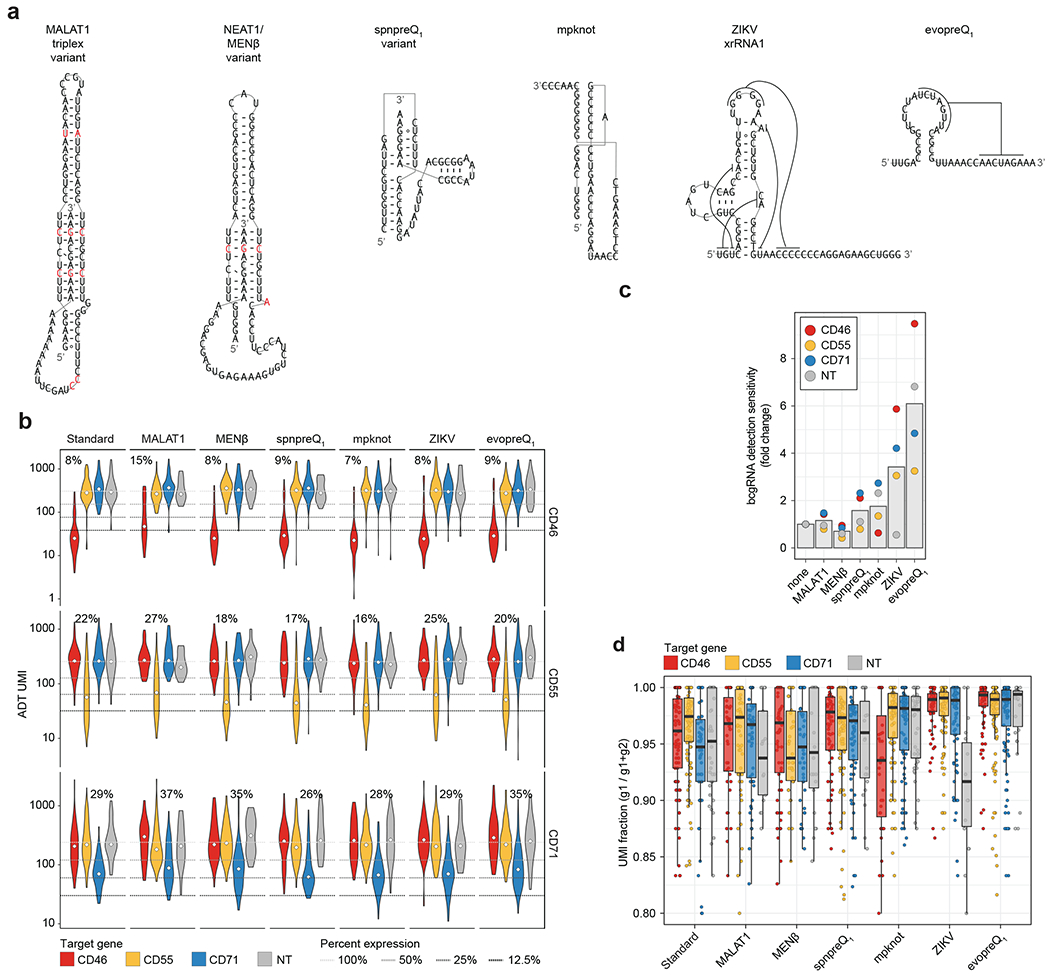
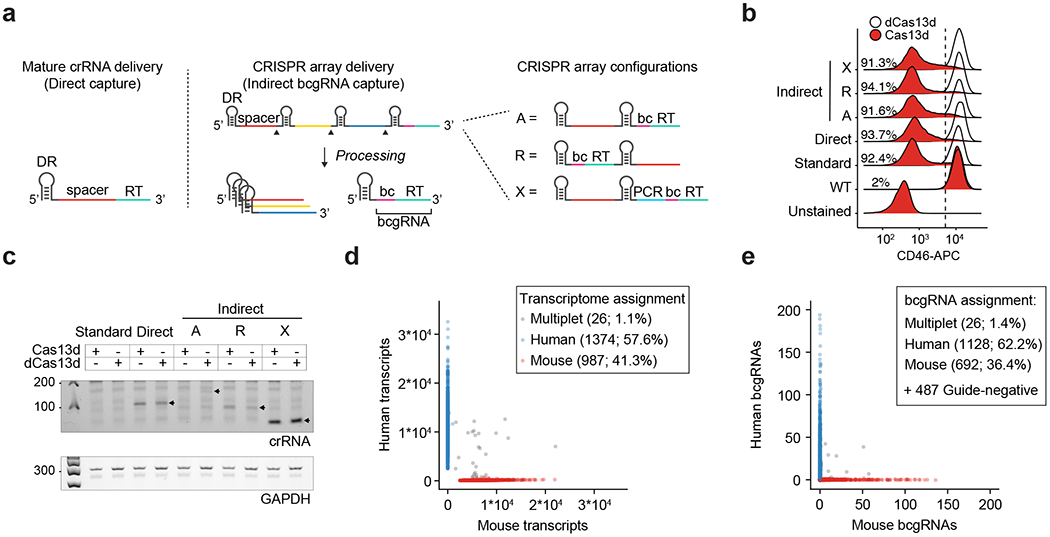
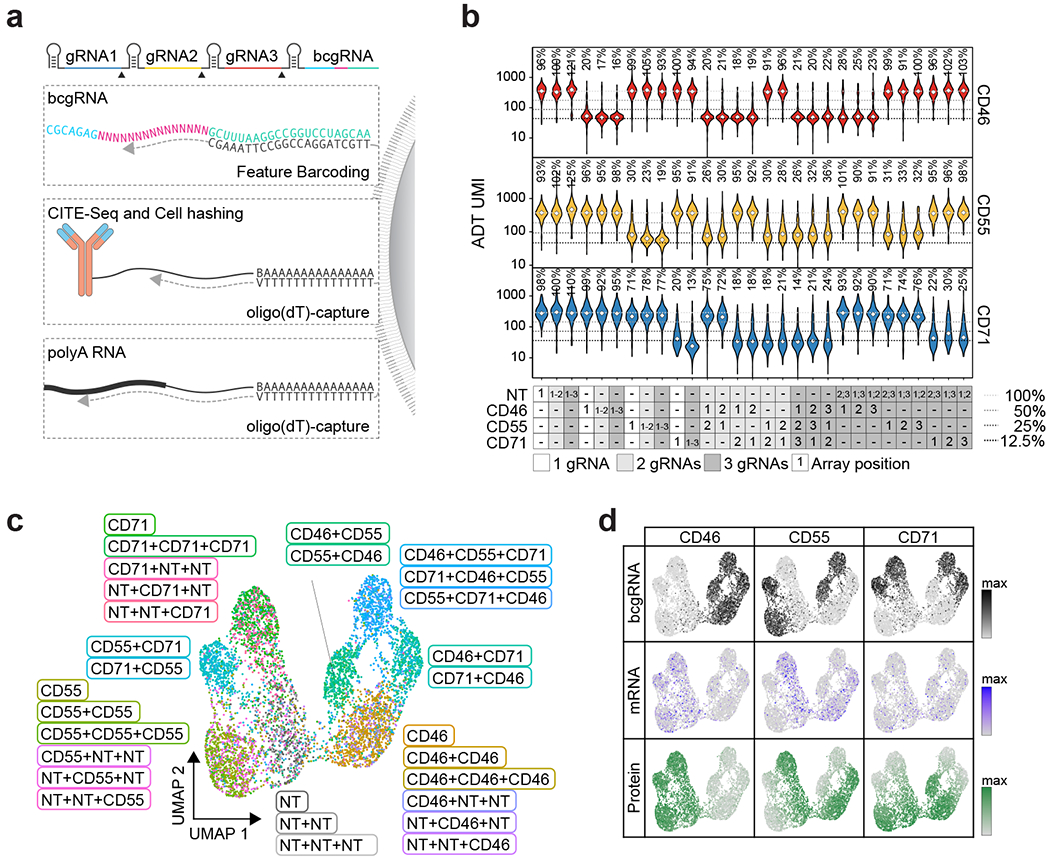
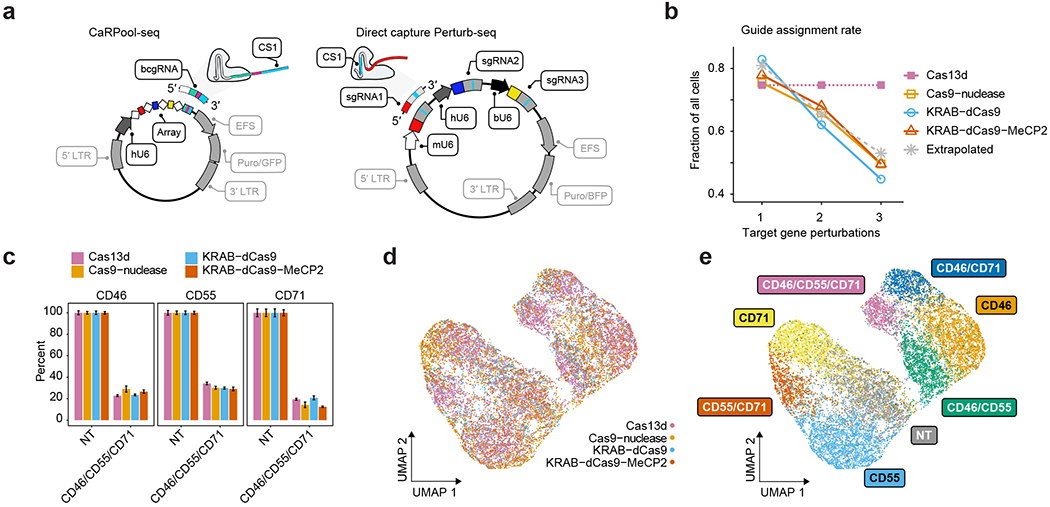
References
-
- Jaitin DA et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167, 1883–1896 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
