

Monitoring the 5'UTR landscape reveals isoform switches to drive translational efficiencies in cancer
- PMID: 36550360
- PMCID: PMC9957725
- DOI: 10.1038/s41388-022-02578-2
Monitoring the 5'UTR landscape reveals isoform switches to drive translational efficiencies in cancer
Abstract
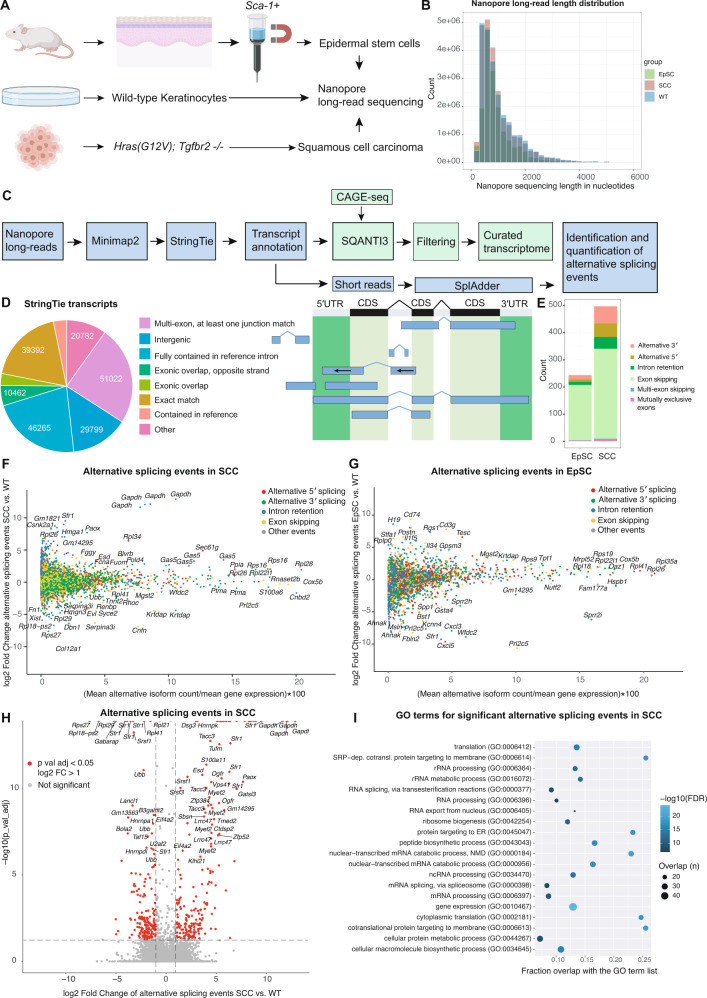
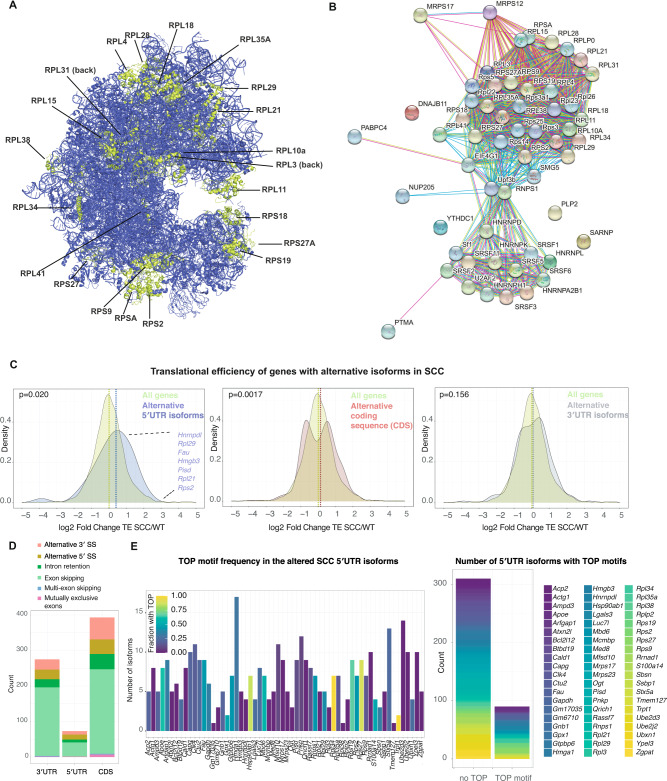
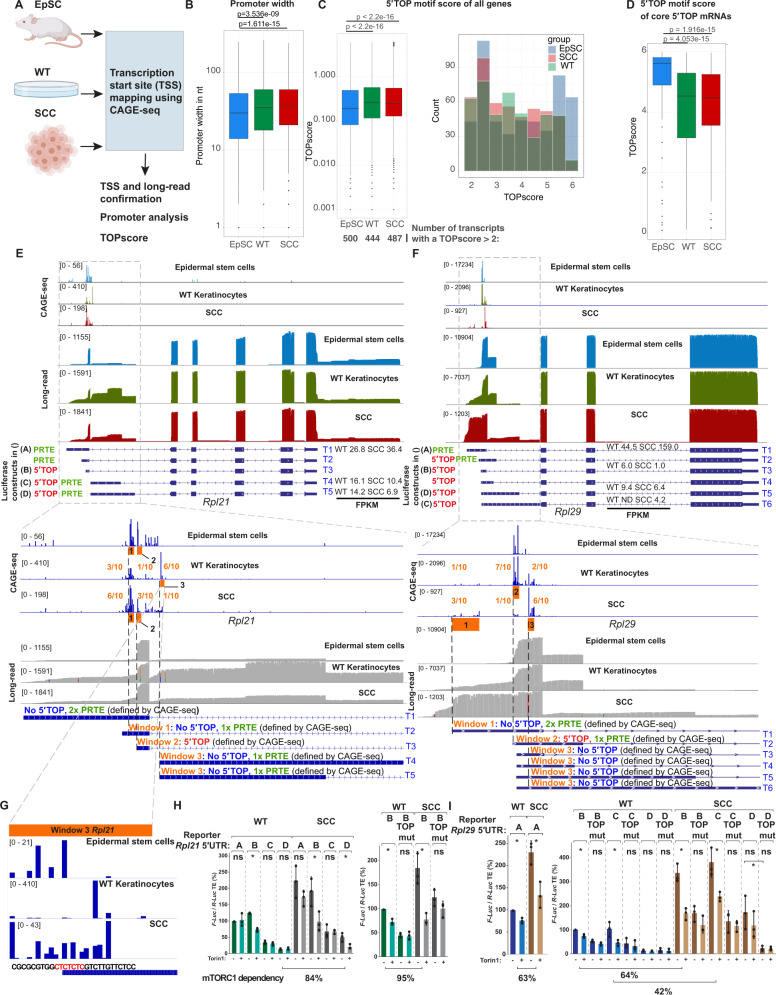
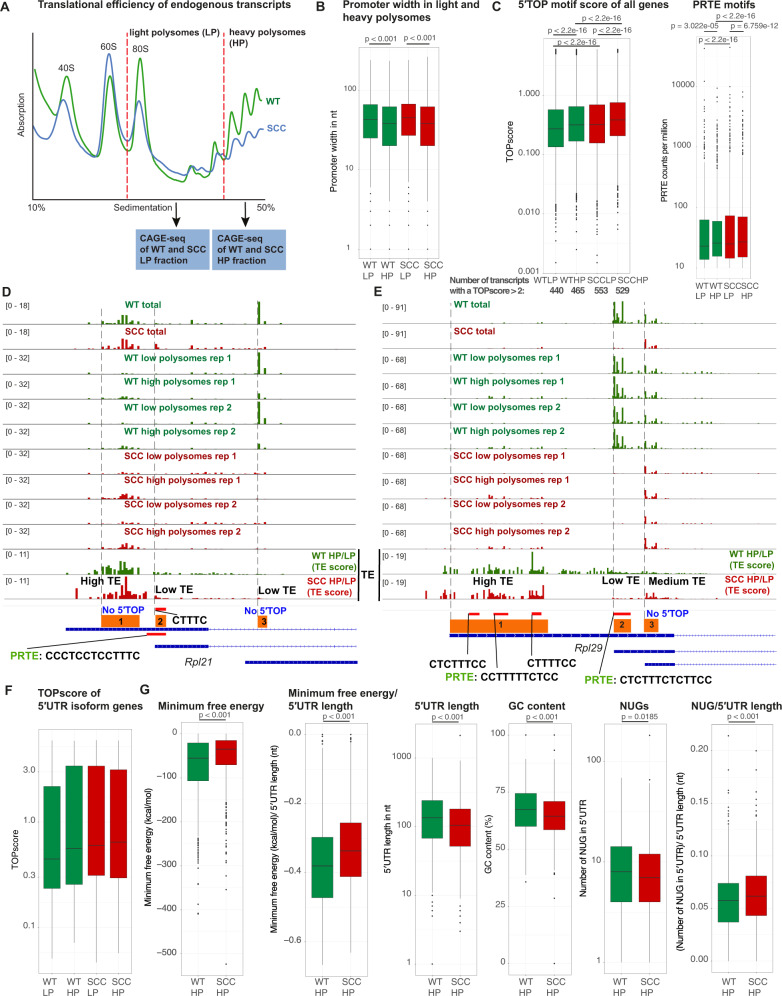
Transcriptional and translational control are key determinants of gene expression, however, to what extent these two processes can be collectively coordinated is still poorly understood. Here, we use Nanopore long-read sequencing and cap analysis of gene expression (CAGE-seq) to document the landscape of 5' and 3' untranslated region (UTR) isoforms and transcription start sites of epidermal stem cells, wild-type keratinocytes and squamous cell carcinomas. Focusing on squamous cell carcinomas, we show that a small cohort of genes with alternative 5'UTR isoforms exhibit overall increased translational efficiencies and are enriched in ribosomal proteins and splicing factors. By combining polysome fractionations and CAGE-seq, we further characterize two of these UTR isoform genes with identical coding sequences and demonstrate that the underlying transcription start site heterogeneity frequently results in 5' terminal oligopyrimidine (TOP) and pyrimidine-rich translational element (PRTE) motif switches to drive mTORC1-dependent translation of the mRNA. Genome-wide, we show that highly translated squamous cell carcinoma transcripts switch towards increased use of 5'TOP and PRTE motifs, have generally shorter 5'UTRs and expose decreased RNA secondary structures. Notably, we found that the two 5'TOP motif-containing, but not the TOP-less, RPL21 transcript isoforms strongly correlated with overall survival in human head and neck squamous cell carcinoma patients. Our findings warrant isoform-specific analyses in human cancer datasets and suggest that switching between 5'UTR isoforms is an elegant and simple way to alter protein synthesis rates, set their sensitivity to the mTORC1-dependent nutrient-sensing pathway and direct the translational potential of an mRNA by the precise 5'UTR sequence.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
