

Alterations of Cytoskeleton Networks in Cell Fate Determination and Cancer Development
- PMID: 36551290
- PMCID: PMC9775460
- DOI: 10.3390/biom12121862
Alterations of Cytoskeleton Networks in Cell Fate Determination and Cancer Development
Abstract
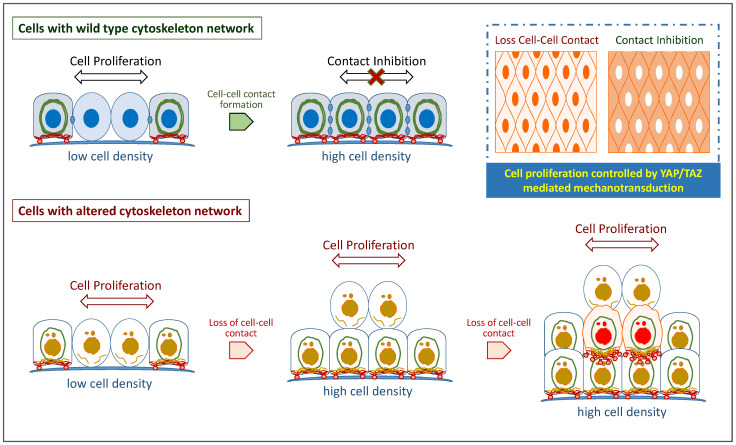
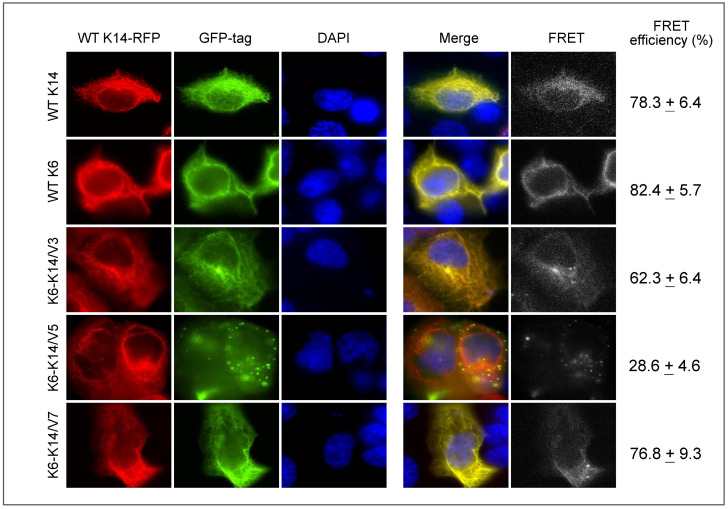
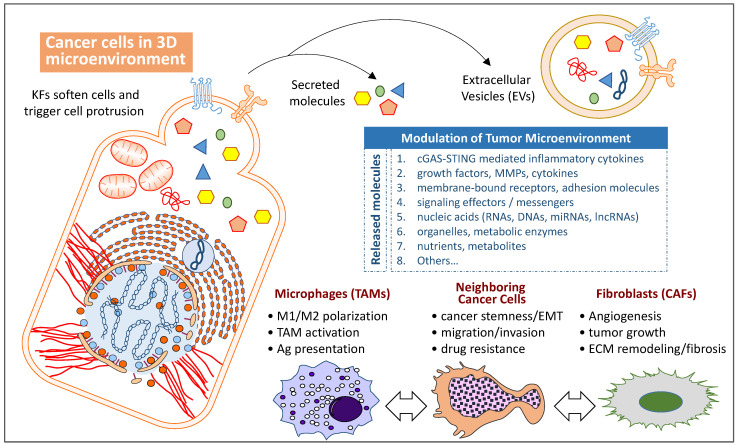
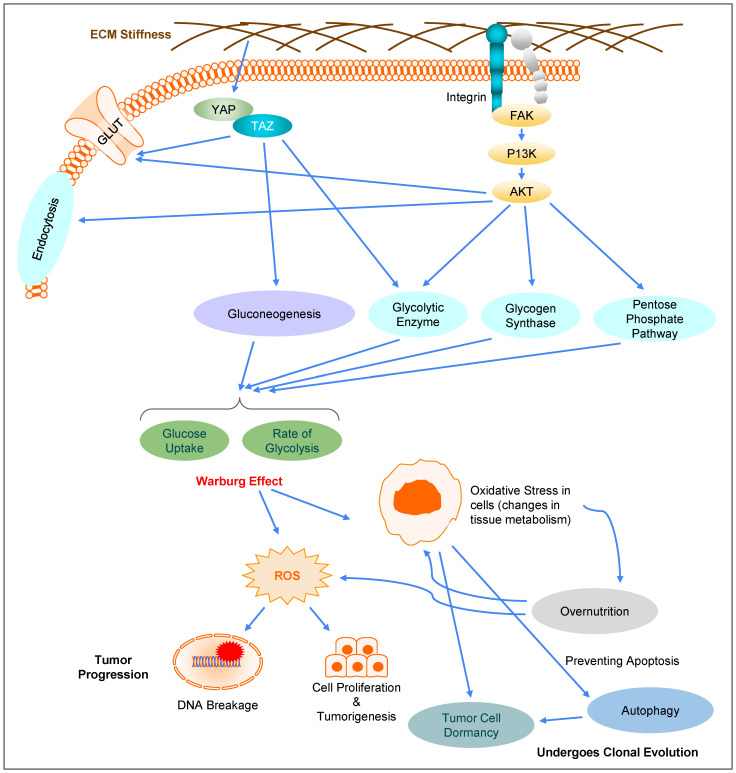
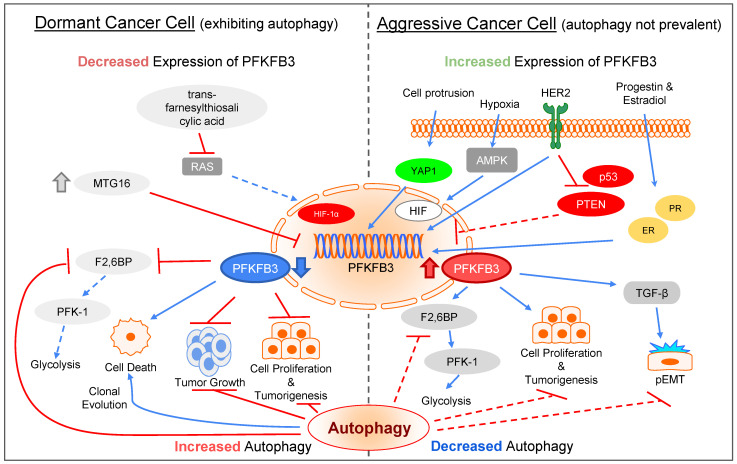
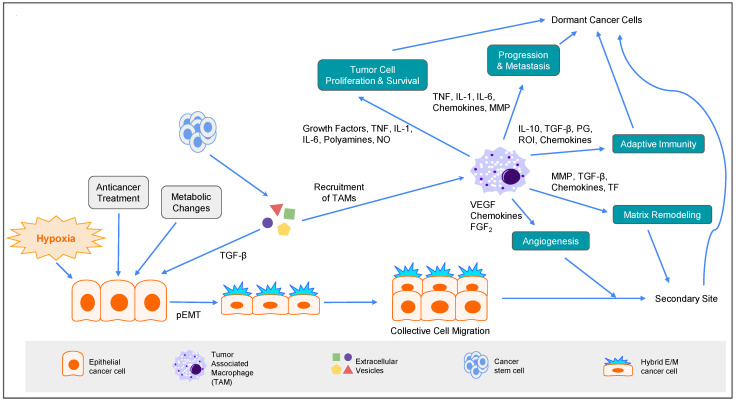
Cytoskeleton proteins have been long recognized as structural proteins that provide the necessary mechanical architecture for cell development and tissue homeostasis. With the completion of the cancer genome project, scientists were surprised to learn that huge numbers of mutated genes are annotated as cytoskeletal or associated proteins. Although most of these mutations are considered as passenger mutations during cancer development and evolution, some genes show high mutation rates that can even determine clinical outcomes. In addition, (phospho)proteomics study confirms that many cytoskeleton-associated proteins, e.g., β-catenin, PIK3CA, and MB21D2, are important signaling mediators, further suggesting their biofunctional roles in cancer development. With emerging evidence to indicate the involvement of mechanotransduction in stemness formation and cell differentiation, mutations in these key cytoskeleton components may change the physical/mechanical properties of the cells and determine the cell fate during cancer development. In particular, tumor microenvironment remodeling triggered by such alterations has been known to play important roles in autophagy, metabolism, cancer dormancy, and immune evasion. In this review paper, we will highlight the current understanding of how aberrant cytoskeleton networks affect cancer behaviors and cellular functions through mechanotransduction.
Keywords: autophagy; cancer; cytoskeleton; dormancy; immune evasion; mechanotransduction; metabolism reprogramming; tumor microenvironment.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
